BASIC ULTRAVIOLET RADIATION PHOTOBIOLOGY
Kendric C. Smith
Emeritus Professor, Radiation Oncology (Radiation Biology)
Stanford University School of Medicine
800 Blossom Hill Road, Unit R169, Los Gatos, CA 95032
kendric@stanford.edu
web.stanford.edu/~kendric/
Introduction
Ultraviolet (UV) radiation is generally divided into three regions, especially in Photomedicine, i.e., UV-C (100-280 nm), UV-B (280-320 nm), and UV-A (320-400 nm). Virtually no UV-C radiation reaches the earth, because of strong filtering by the ozone layer, however, most experiments on the effects of UV radiation on cells are performed using UV-C (e.g., 254 nm), because of it efficiency in producing damage to cells, especially to their deoxyribonucleic acid (DNA).
While research on UV radiation is largely focused on its detrimental effects, UV radiation also has a very important beneficial effect, i.e., the production of Vitamin D in the skin of humans. Vitamin D3 is produced photochemically in the skin from 7-dehydrocholesterol. The highest concentrations of 7-dehydrocholesterol are found in the epidermal layer of skin. Synthesis in the skin involves UV-B radiation (280-320 nm), which effectively penetrates only the epidermal layers of skin. While 7-dehydrocholesterol absorbs UV radiation at wavelengths between 270-300 nm, optimal synthesis occurs in a narrow band between 295-300 nm (Vitamin D).
This module, however, will focus on the detrimental effects of UV radiation, and the many mechanisms that cells have for repairing this damage. These same repair systems are also very important for repairing metabolic damage to DNA, i.e., produced just from normal living and breathing.
Lethal and Mutagenic Effects of UV Radiation
UV radiation produces many types of photochemical alterations in DNA, ribonucleic acid (RNA), and protein, as well as to structures such as membranes. DNA, however, is the major target for the deleterious effects of UV radiation because it is the largest molecule in the cell, it is present in the fewest copies, it carries the genetic information for a cell, and it absorbs UV radiation very efficiently. In contrast, RNA and proteins are present in multiple copies, so that it is much more difficult to inactivate a significant number of these molecules in a cell by UV irradiation.
A large number of different types of damage are produced in DNA by UV irradiation. These include the modification of individual purine and pyrimidine bases (e.g., deamination, ring cleavage), the production of pyrimidine dimers (i.e., the covalent linkage of adjacent pyrimidines in DNA), and the addition of other molecules to the purines and pyrimidines (e.g., water (photohydrate), DNA-Protein Crosslinks.).
The direct action of UV radiation does not produce strand breaks in DNA (but X-rays do), but single and double-strand breaks in DNA are produced as a by-product of DNA repair (see module on DNA Double-Strand Breaks).
With photosensitized reactions, near-UV radiation or visible light activates a sensitizer; the sensitizer can then combine with DNA (e.g., psoralen, as in the treatment of psoriasis), or can transfer its energy to a new molecule. These reactions are highlighted in the modules on Photosensitization.
While the most drastic effect of UV radiation on a cell is lethality, UV radiation-induced damage can also result in mutations, i.e., alterations in the DNA base sequence of a gene such that the capacity of the gene product to perform an essential cellular function is altered. Some mutations can be lethal (see module on UV Radiation and Spontaneous Mutagenesis).
Fortunately, a cell has an enormous capacity to repair all types of damage to its DNA. These repair systems have names like photoreactivation, base excision repair, nucleotide excision repair, recombinational repair, and double-strand break repair (discussed below).
These DNA repair systems have not evolved just to repair damage produced by UV radiation, they are also required to maintain the integrity of the DNA after damage by the products of normal metabolism (e.g., superoxide), and to repair the heat damage produced at body temperature (e.g., deamination). Some of these repair systems are very accurate, and do not produce mutations. Other systems, however, are efficient, but inaccurate, and do produce mutations.
The first indication that cells could recover from UV radiation-induced damage was the observation by Roberts and Aldous in 1949 that minor modifications in the postirradiation treatment of certain bacterial strains resulted in a marked increase in their viability. Subsequently, it was observed that UV irradiated E. coli rec- cells showed enhanced survival when held under non-growth conditions for various periods of time before plating on growth medium to assay for survival. This process is called "liquid holding recovery". Thus, the delay of macromolecular synthesis (i.e., DNA, RNA, and protein) for a time after irradiation led to a higher survival. By holding cells in non-growth medium, it allows the cells to complete constitutive repair processes, e.g., nucleotide excision repair, without interference with the inducible recombinational repair systems in these repair-impaired Rec strains.
The ultimate proof that cells could recover from radiation damage was the isolation in 1958 of a radiation sensitive mutant of Escherichia coli (Bs-1) by Ruth Hill. This discovery inaugurated the era of the genetic approach to the study of DNA repair and mutagenesis.
Genetic studies have been responsible for most of the discoveries about the biochemical basis of DNA repair and mutagenesis. For example, one can measure the initial yield of a particular type of radiation-induced lesion in the DNA of a wild-type bacterial strain (i.e., radiation resistant) and in a mutant strain (i.e., radiation sensitive), and then follow the kinetics and extent of the repair of that lesion, and thereby determine if the mutation under investigation has an effect on the repair of that type of DNA lesion.
Then by constructing strains that carry two mutations that affect DNA repair, one can determine through survival and DNA repair studies whether the two mutations act in the same or in different biochemical pathways for repair. For example, since the uvrA recA double mutant of E. coli is much more sensitive than either of the singly mutant strains, it indicates that the uvrA and recA genes function in separate pathways of DNA repair (Figure 1).
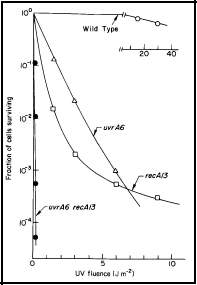
Figure 1. UV radiation survival curves for DNA repair deficient mutants of E. coli K-12. The uvrA6 mutation blocks nucleotide excision repair, and the recA13 mutation blocks recombinational DNA repair. Note that the double mutant, uvrA6 recA13 is very much more sensitive to UV radiation than either of the single mutants, indicating that the two single mutants are involved in separate pathways of DNA repair. Since the two single mutants have about the same sensitivity, it indicates that nucleotide excision repair and recombinational DNA repair are of about equal importance to the survival of UV irradiated E. coli. [Modified from Howard-Flanders and Boyce, 1966]
The uvrA mutation blocks nucleotide excision repair, a repair system that is largely constitutive and error free. The recA mutation blocks recombinational repair, which is in part inducible by radiation damage, and therefore requires protein synthesis for it to work. This type of repair has a component that is error prone, i.e., it is efficient but makes mistakes, and thus leads to mutations. These two DNA repair systems are sometimes called "dark repair" systems, since they do not need the presence of light for them to function.
Photoreactivation: There is a type of repair that requires light to function, and is found in a wide range of organisms. Thus, if E. coli are exposed to a lethal dose of UV radiation (e.g., 254 nm), lethality can be reduced or eliminated by a second exposure to light in the range of 300-450 nm. This process requires an enzyme, DNA photolyase (a flavoprotein), which binds to pyrimidine dimers. When this enzyme-substrate complex absorbs light in its flavine moiety at the above wavelengths, the pyrimidine dimer is split yielding undamaged pyrimidines, and the enzyme is released (Sancar, 2000). [see module on Photoreactivation]
Nucleotide Excision Repair: In this process (Figure 2), a distortion in the DNA caused by a lesion (e.g., pyrimidine dimer) is recognized by the UvrABC nuclease. It cuts on both sides of the lesion, producing a gapped structure that is about 12 nucleotides long. Repair replication, using DNA polymerase I, fills in the gap using the undamaged opposite strand of DNA as the template. Finally the break in the repaired DNA strand is sealed by DNA ligase (Petit and Sancar, 1999). This repair system is largely constitutive, and is error free.
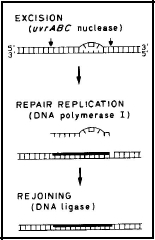
Figure 2. Nucleotide Excision Repair in E. coli. The UvrABC nuclease recognizes the distortion in the DNA due to the lesion, then cuts on both sides of the lesion, producing a gapped structure about 12 nucleotides long. Repair replication (heavy line) fills the gap using the strand opposite the gap as the template. Finally, the break in the strand is sealed by DNA ligase. [Modified from Sancar and Rupp, 1983]
For a discussion of nucleotide excision repair in mammalian cells, see the module Nucleotide Excision Repair in Human Cells.
Base Excision Repair: Altered purine and pyrimidine bases (e.g., thymine glycol, 3-methyladenine, uracil, urea) are recognized by a class of enzymes called DNA glycosylases, which are specific to varying degrees for particular types of altered bases. These enzymes split off the altered base at the N-glycosylic bond that links the base to the sugar in the DNA backbone. This site of the base loss is called an AP site (for apurinic or apyrimidinic). The AP site is degraded to produce a gap in the DNA strand, which is repaired by the same processes used to repair the gaps during nucleotide excision repair. This process is especially important for cells exposed to ionizing radiation and alkylating agents, but is also important for the repair of normal metabolic damage, including depurination at normal body temperature (Seeberg et al., 1995).
Recombinational Repair: This type of repair is much more complicated than is excision repair, and requires many more gene products. The products of a number of these repair genes are induced by radiation damage, and therefore this type of repair requires protein synthesis before it can function. Because of its complexity, this type of repair makes mistakes (Figure 3).
Recombinational repair requires four strands of DNA (i.e., two sister duplexes). One type of recombinational repair is called Postreplication Repair, and comes into play after replication proceeds past a lesion in DNA, leaving a gap in the replicated strand opposite the lesion. This gap is then filled with material with the correct base sequence from the sister duplex strand. During this process of strand exchange from one sister duplex to another, however, slippage in the base pairing of these DNA strands can occur, leading to mutations, which can be lethal (Rupp et al., 1971; Howard-Flanders and Rupp, 1972; Smith, 2004; Kuzminov, 1999). [see module on Recombinational DNA Repair]
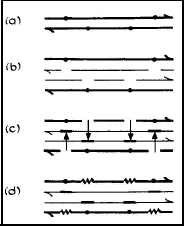
Figure 3. Postreplication Repair (recombinational DNA repair) in UV irradiated E. coli. (a) The dots indicate lesions in the DNA. (b) DNA synthesis proceeds up to a lesion and then skips past the lesion, leaving a gap in the daughter strand. (c) Filling of the daughter strand gaps with DNA from parental strands by a recombinational process that requires a functional recA gene. (d) Gaps in the parental strands are repaired by repair replication (see Nucleotide Excision Repair). [Modified from Smith, 1989]
Recombinational Excision Repair: This process occurs in the part of the chromosome that was replicated prior to irradiation, i.e., where two sister duplexes were present before irradiation. After the excision of the lesion, the resulting gap is filled by the same recombinational process just described for postreplication repair (Figure 4).
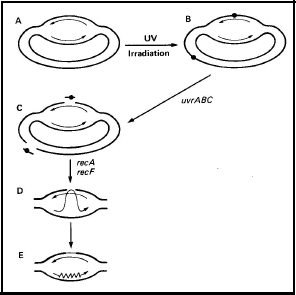
Figure 4. The recombinational repair of excision gaps in E. coli. UV radiation-induced lesions are produced in both the replicated and unreplicated portions of the genome (B). The gaps produced by excision in the unreplicated portion are repaired by the classical methods of nucleotide excision repair (C). The gaps produced in the replicated portion of the chromosome are repaired by a recombinational process that requires both recA and recF (C-D). [Modified from Smith and Sharma, 1987]
Comclusions
Each of these "dark" repair systems involve the cutting of DNA strands, and this can often lead to the formation of DNA double-strand breaks, which are lethal if not repaired (see module on DNA Double-Strand Breaks). Fortunately, cells are very good at repairing DNA double-strand breaks by recombinational processes of the type just described, i.e., requiring the presence of two sister duplexes (Bonura and Smith, 1975a, b). These processes are called homologous recombination, since "proper" base pairing is involved.
Some cells can also have the capacity for non-homologous recombination (non-homologous end joining), where the ends of a double-strand break are simply sealed together, which usually results in the loss of bases, and therefore results in lethality or mutation.
Mutations are not only produced by errors in repair, they can also be produced by errors made during normal DNA synthesis. This was first suggested by the finding that some mutations are produced independent of the recombinational repair systems. There are special systems for the prevention, detection, and repair of errors made during DNA replication (e.g., Mismatch Repair).
Most of our early knowledge about the biochemistry and genetic control of the multiple pathways of DNA repair has come from studies on bacteria, especially E. coli, however, when the granting agencies decreed that people should work on human cells in order to obtain funding, the beautiful and not nearly complete work on bacteria essentially ceased. Then there were a few years of slow progress as people switched to eucaryotic cells (yeast and human cells). At that time, there were only a few mutants available for these cells, and the culturing of these cells is much more difficult and slower than for bacteria. Nevertheless, great progress has been made in our understanding of DNA repair and mutagenesis in both yeast and human cells.
Mammalian cells have essentially the same repair systems that were first discovered in bacteria, however, repair is more complicated in mammalian cells because the structure of the genome is more complicated. In bacteria, the DNA is essentially naked, while the DNA in mammalian cells is surrounded by proteins. These proteins must first be removed before DNA repair can proceed. Therefore, more genes are involved in nucleotide excision repair in mammalian cells than in bacteria (see module on Nucleotide Excision Repair in Human Cells).
DNA repair mutants in yeast can be produced in a manner similar to the production of mutants in bacteria, i.e., treat the cells with a mutagen and then select for the phenotype (characteristic) that you want, i.e., sensitivity to radiation. The mutants in human cells have largely come from patients who have shown a sensitivity to sunlight (e.g., Xeroderma pigmentosum) or to ionizing radiation, detected during radiation therapy for cancer (e.g., Ataxia telangectasia).
Through the use of such mutant cells, the pathways of DNA repair in mammalian cells have been discovered and studied in the same general manner that was used in the bacterial studies.
References
NOTE: The author's papers cited here are available as PDF files at: web.stanford.edu/~kendric/
Bonura T, Smith KC. 1975a. Enzymatic production of deoxyribonucleic acid double-strand breaks after ultraviolet irradiation of Escherichia coli K-12. J Bacteriol 121:511-517.
Bonura T, Smith KC. 1975b. Quantitative evidence for enzymatically-induced DNA double-strand breaks as lethal lesions in UV-irradiated pol+ and polA1 strains of E. coli K-12. Photochem Photobiol 22:243-248.
Howard-Flanders P, Boyce RP. 1966. DNA repair and genetic recombination: studies on mutants of Escherichia coli defective in these processes. Radiat Res Suppl 6:156.
Howard-Flanders P, Rupp WD. 1972. Recombination repair in UV-irradiated Escherichia coli, In: Molecular and Cellular Repair Processes (Beers Jr. RF, Herriott RM, Tilghman RC. ed), Johns Hopkins Medical Journal, Suppl. 1, p 212-225.
Kuzminov A. 1999. Recombinational repair of DNA damage in Escherichia coli and bacteriophage lambda. Microbiol Mol Biol Rev 63:751-813.
Petit C, Sancar A. 1999. Nucleotide excision repair: from E. coli to man. Biochimie 81:15-25.
Rupp WD, Wilde III CE, Reno DL, Howard-Flanders, P. 1971. Exchanges between DNA strands in ultraviolet-irradiated Escherichia coli. J Mol Biol 61:25-44.
Sancar GB. 2000. Enzymatic photoreactivation: 50 years and counting. Mutat Res. 451(1-2):25-37.
Sancar A, Rupp W.D. 1983. A novel repair enzyme: UVRABC excision nuclease of Escherichia coli cuts a DNA strand on both sides of the damaged region. Cell 33,249-260.
Seeberg E, Eide L, Bjoras M. 1995. The Base Excision Repair Pathway. Trends Biochem Sci. 10:391-397.
Smith KC. 1989. UV Radiation Effects, in The Science of Photobiology, 2nd edition, (Smith KC, ed), Plenum Press, NY., pp.111-134.
Smith KC. 2004. Recombinational DNA repair: the ignored repair systems. BioEssays 26:1322-1326.
Smith KC, Sharma RC. 1987. A model for the recA-dependent repair of excision gaps in UV-irradiated Escherichia coli. Mutat Res 183:1-9.
09/17/08
03/29/09
06/02/11
03/19/14