NUCLEOTIDE EXCISION REPAIR in HUMAN CELLS
James E. Cleaver
Auerback Melanoma Laboratory
Box 0808, UCSF Cancer Center, University of California
San Francisco, CA 94143-0808
james.cleaver@ucsf.edu
Introduction
Nucleotide excision repair (NER) is a versatile process that can remove many forms of DNA damage by nuclease cleavage on either side of the damaged bases, removal of the damaged oligonuclotide, and resynthesis of a patch using the undamaged strand as the template. This apparently simple scheme has been known in outline for many years, but its intricacies continue to amaze and provide employment for many investigators. Understanding the intricacies requires examination of the various conceptual steps involved including (a) substrate specificity, (b) damage recognition, (c) unwinding, (d) excision, and (e) resynthesis. Although the reconstruction of NER with individual proteins in vitro has provided major insights into the mechanisms of repair, there may be significant differences to the details when the substrate is compact intranuclear chromatin.
In cells, NER can also be the initiator of a series of signal transduction processes involving the kinases ATM (ataxia telangiectasia mutated) and ATR (ataxia telangiectasia and Rad3-related), which transduce damage and repair into cell cycle delays, chromosome aberrations, genomic instability, apoptosis and senescence through phosphorylation of many downstream protein substrates (Abraham, 2001; Falck et al., 2005; Hurley and Bunz, 2007; Matsuoka et al., 2007; Wang et al., 2005). Several components of NER also have additional roles in ubiquitination, telomere maintenance, DNA replication, and transcription or gene expression. Mutations in NER genes are associated with several human diseases, xeroderma pigmentosum (XP), Cockayne Syndrome (CS) and trichothiodystrophy (TTD), which exhibit increased cancer incidence, developmental delay and neurodegeneration (see module on Human Photosensitive Diseases of DNA Repair).
Substrates For NER
The NER system recognizes and repairs DNA damage of a very wide range of chemical structures, from small oxidative modifications to large DNA adducts. Much of the initial development of NER was based on the repair of the UV-induced photoproducts. The two major photoproducts are cyclobutane pyrimidine dimers (CPDs) and at about 25% the frequency, [6-4]-pyrimidine dimers ([6-4]PDs), which are preferentially induced at thymine-cytosine dipyrimidines (Mitchell and Cleaver, 1990). Cytosine absorbs longer wavelengths of UV more efficiently than thymine, and therefore CPDs containing cytosine are formed more readily after UVB (280-320 nm) irradiation (Ellison and Childs, 1981), and may play a major role in UVB (solar) mutagenesis. The [6-4]PD can undergo a UVB-dependent conversion to its valence photoisomer, the Dewar pyrimidinone (Taylor and Cohrs, 1987). The distribution of photoproducts in DNA is influenced by chromatin structure, e.g., higher relative amounts of [6-4]PDs are found in linker DNA between nucleosomes as compared to the core DNA (Mitchell et al., 1990b). Chemical adducts include those produced by carcinogens or chemotherapy agents such as N-acetoxy-N-acetyl aminofluorene, benzo(a)pyrene, aflatoxin, photoactivated psoralens, and cisplatin. The neurological degeneration seen in some XP, CS and TTD patients may be due to unrepaired damage generated endogenously by reactive oxygen species (Cleaver and Revet, 2008; Laposa et al., 2007), abnormalities in mitochondrial function (Scheibye-Knudsen et al., 2013) or downstream disruption of gene expression (Tresini et al., 2015).
There is considerable variation in the efficiency with which these various forms of DNA damage are recognized by the NER system (Figure 1). The efficiency may be affected by the degree to which the damage itself causes intrinsic loss of hydrogen bonding. Using a cell-free system, [6-4]PDs and cisplatin adducts are the most readily detectable of all lesions, with CPDs excised at about 20% the efficiency, and other lesions with lower efficiencies (Reardon and Sancar, 2005). An oxidative lesion (8-oxoG) is recognized as efficiently as CPDs. The in vitro assay detected the apparent repair of undamaged DNA (see Figure 1 "UM") at about 1% of the level of [6-4]PDs; this could be real untargeted repair or the background activity of the assay. The difference between the efficiencies for [6-4]PD and CPD repair in vitro explains a large part of the differences between the rates of excision of these photoproducts seen in live cells (Mitchell et al., 1985).
In cells, additional factors play a role in repair, such as chromatin structure, and the activity of proteins that were missing from the in vitro reactions (e.g., DDB1/2, p53). Consequently the differences in the rates of excision are even greater than the in vitro efficiencies would predict. In human cells, on average, the half time for removal of [6-4]PDs is about 1 hr, and CPDs approximately 12-24 hr. CPD repair is, in addition, more rapid from transcribed regions of the genome than from nontranscribed regions. In rodent cells, the difference is even greater, with rapid removal of [6-4]PDs, but almost no CPD removal from genomic DNA (Klimek, 1965; Mitchell et al., 1990a ,1999). The loss of transcriptional repair factors in CS results in preferential loss of CPD repair, rather than [6-4]PD repair (Barrett et al., 1991; Parris and Kraemer, 1993). The re-expression of XPC DNA damage binding protein preferentially restores CPD repair over [6-4]PD (Emmert et al., 2000). These studies suggest that the repair of CPDs in vivo is a much greater challenge to the NER system than larger or more distorting forms of damage, and may require the activity of more gene products than does the repair of [6-4]PDs.
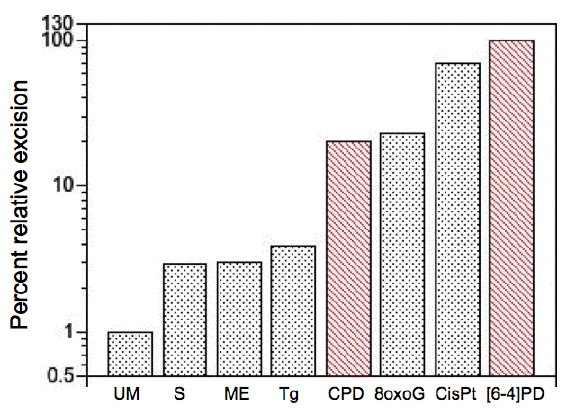
Figure 1. Relative levels of excision by the mammalian excision nuclease system. Chinese hamster ovary cell-free extracts were used under substrate-limiting conditions. Lesions include undamaged (unmodified, UM) thymidine with a subset of backbone and base alterations that are substrates for the E. coli and mammalian excision nucleases. Phosphorothioate (S)- and methylphosphonate (ME)-adducted thymidines cause minor alterations in the sugar-phosphate backbone. Thymine glycol (Tg) and 8-oxoguanine (8oxoG) are generated by reactive oxygen species, and cause minor helical distortions. Exogenous agents introduce bulky, helix-distorting lesions, such as CPD and [6-4]PD generated by UV radiation, and the 1,2-d(GpG) intrastrand diadduct (CisPt) is introduced by the chemotherapeutic agent cisplatin. The average percentage excision observed for [6-4]PD (10%) was defined as 100, and other percentages were normalized relative to this value. [Redrawn from Reardon and Sancar (2005)]
Due to the more rapid excision of [6-4]PDs than CPDs, many early measurements of repair that were made during the initial hours after irradiation (unscheduled DNA synthesis, repair replication, strand breakage) were biased towards these photoproducts rather than CPDs (Cleaver and Thomas, 1981; Erixon and Ahnstrom, 1979; Painter and Cleaver, 1969; Tang et al., 1986). This led to early confusion, because the measures of repair during the first hours after UV irradiation did not correlate with the measures of CPDs. Fortunately the appreciation that UV radiation makes several photoproducts with different excision rates has resolved these issues.
Biochemistry
The NER system consists of a sequential series of reactions involving a large number of proteins (Figure 2), by which DNA damage is recognized in nuclear DNA within different functional domains, and subsequently excised and replaced (Table 1) (Hoeijmakers, 2001; Wood, 2001).
Table 1. Names, functions and chromosome location of most nucleotide excision repair genes.

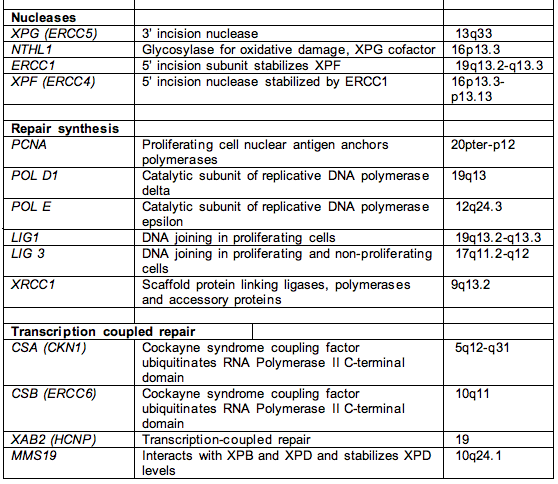

Footnote. This list includes most of the genes involved in NER, except for RNA polymerase I and II and accessory factors, and the signal transduction protein kinases ATM and ATR that transduce damage from NER to hundreds of downstream targets. The proteins involved in the replication of UV-damaged DNA, e.g., Pol eta and iota, represent another large class of proteins not represented in this table.
Damage in transcriptionally inactive regions is detected by interactions between the DNA damage binding proteins DDB1/DDB2(XPE), HR23B/XPC and XPA/RPA. (RPA is replication protein A, also known as single strand binding protein.) Damage in transcriptionally active regions is detected through arrest of the transcriptional machinery involving RNA polymerase I and II (Lindsey-Boltz and Sancar, 2007), and requires the CSA, CSB and UVSSA proteins that are mutated in the human diseases Cockayne Syndrome (CS) and Ultraviolet Sensitive Syndrome (UVS) (Itoh et al., 1995; Cleaver 2012). . This results in an increased rate of repair known as transcription coupled repair (TCR). The damaged site is then remodeled by the concerted action of the DNA binding protein heterodimer XPA/RPA, and the unwinding activity of the XPB and XPD proteins, which are components of a 10 subunit transcription factor TFIIH. Cleavage then occurs on both sides of the damaged site: first 22 nucleotides 5' to the dimer by the XPF/ERCC1 5' nuclease, and then 5 nucleotides 3’ to the dimer by the cryptic XPG nuclease that cleaves once polymerization of the patch by DNA polymerase reaches its site of binding (Fagbemi et al., 2011). These sites may vary according to the precise nature of the damage, and the excised oligonucleotide can be between 24 and 32 nucleotide in length. The patch is resynthesized by a polymerase anchored to the replication site by proliferating cell nuclear antigen (PCNA) and sealed by a ligase.
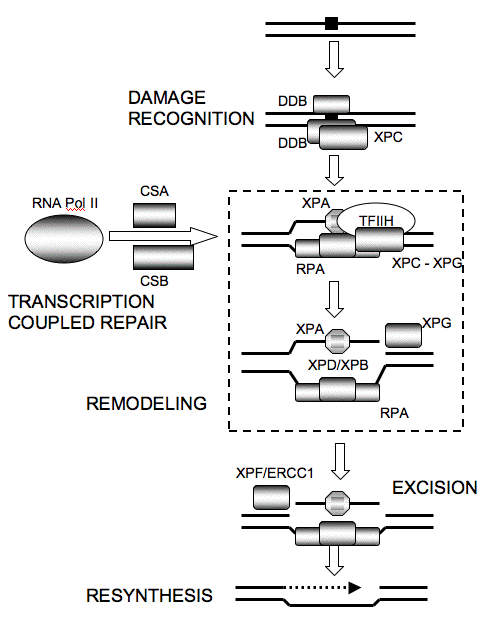
Figure 2. Nucleotide Excision Repair as a Linear Sequence of Reactions. Damage recognition for global repair occurs via DDB1/DDB2(XPE), and HR23B/XPC. Damage recognition for transcription coupled repair occurs via RNA Pol II arrest and the coupling factors CSA and CSB. XPC interacts with TFIIH, facilitating unwinding by the XPB and XPD members of TFIIH. Remodeling via the 10 component transcription factor TFIIH allows XPA/RPA to enter the open complex. RPA is replication protein A, and is also known as single-strand binding protein. XPG is downloaded through interaction with TFIIH to cleave 3' to the dimer, XPF/ERCC1, which interacts with XPA, and cleaves first on the 5' side. The patch is resynthesized by polymerase, PCNA and ligase.
Damage Recognition
The recognition process for DNA damage in human cells is considerably more complex than in E. coli. The recognition proteins include the heterodimers DDB1/DDB2(XPE), XPC/HR23B, and XPA/RPA, each of which have binding activity on damaged DNA. DDB1/DDB2(XPE) is not needed in vitro, and mutations in XPE result in only a mild XP disease, and small reductions of about 50% in the excision of CPDs (Zelle and Lohman, 1979). The binding sites for DDB1/DDB2(XPE) on damaged DNA encompasses both DNA strands and masks the 3' cleavage site, so it must be displaced before this cleavage can occur (Figure 3) (Reardon et al., 1993). The complex ubiquitinates XPC, which increases the binding capacity of XPC on damaged DNA (Sugasawa et al., 2005), and stimulates excision (Tang et al., 2000). The promoter of XPE has p53 response elements and its expression increases upon UV irradiation; in rodent cells, however, the promoter is mutated or methylated, and is no longer subject to p53 transactivation (Tang and Chu, 2002). Even so, in rodents, DDB1/DDB2(XPE) may have additional functions that are in parallel with XPC, because double homozygote mice Xpe+.Xpc+ are inviable (Hoeijmakers, personal communication). XPC expression is regulated by p53 (Ford, 1998), and both XPE and XPC are regulated by vitamin D (Moll et al., 2007).
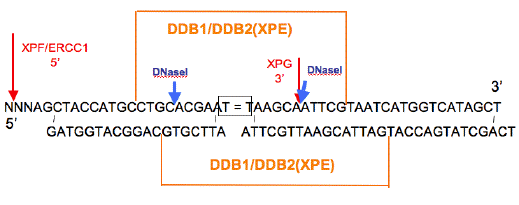
Figure 3. Diagram of DNase I Footprints of DDB1/DDB2(XPE). The adducted thymines are boxed. The orange lines above and below the sequence indicate the areas of protection from DNase I digestion of the top and bottom strands, respectively. Blue arrows indicate DNase I hypersensitive sites. The 3' hypersensitive site for DDB disappears when stoichiometric amounts of XPE (p41) are present. The footprint on the damaged strand is for DNAs containing CPDs, [6-4]PD and [6-4]DewarPD. The footprint for the undamaged strand is for a [6-4]PD containing duplex. [Redrawn from Reardon et al., (1993) to show sites of XPG and XPF/ERCC1 cleavage (red).]
A number of proteins in addition to the core GGR proteins DDB2 and XPC further regulate damage recognition and excision. Within minutes of UV irradiation, DDB2 and PARP1 home onto the photoproducts in DNA. DDB2 and XPC are ubiquitylated by the ligase complex DDB1-DDB2-Cul4A-Rbx1 that increases the affinity of XPC for the damaged site (Robu et al., 2013). PARP1 also adds poly(ADP-ribose) polymers onto the ligase complex. Further modifications facilitate binding and later release of XPC involve SUMOlyation, USP7 ubiquitin protease, arkadia, ALC1 and p97 segregase.
Two approaches have been employed to identify the components and their sequence of action: one is irradiation of subnuclear regions of repair deficient cells, and the observation of the arrival of repair proteins on damaged DNA (Oh et al., 2007; Volker et al., 2001); the other is the reconstitution of the components in vitro using purified native or recombinant proteins (Reardon and Sancar, 2005; Wood, 1999). The primary damage recognition protein is XPC, aided by DDB2 and PARP1. Surprisingly, however, the main DNA binding motif for XPC is on the undamaged strand, such that the CPD is extruded as an extrahelical structure from the other strand (Figure 4) (Maillard et al., 2007). The amino acids Trp690 and Phe733 are essential for the preferential recruitment of XPC protein to its substrates through a high affinity for single-stranded sites. This is a distinctive feature of functional oligonucleotide/oligosaccharide-binding folds found also, e.g., in RPA. In cells that lack XPC, the residual repair occurs in clustered regions of the genome (Kantor et al., 1990; Karentz and Cleaver, 1986; Mansbridge and Hanawalt, 1983). Although XPC cells retain transcription coupled repair, these clustered regions are larger than individual genes, and may represent genomic domains containing multiple actively transcribed genes. XPC is also a critical component of the stem cell coactivator complex that also involves the gene products, NANOG, OCT4, and SOX2 in the pluripotency gene regulatory complex (Zhang et al., 2015)

Figure 4. Versatile Damage Recognition: Detection of Single-Stranded Configurations in the Undamaged Strand of the Double Helix. (A) Initial interaction of XPC protein with damaged sites driven by an affinity for native single-stranded DNA. The triangle symbolizes a helix-distorting bulky lesion. This mechanism with inverted DNA strand specificity directs the XPC protein to the undamaged strand, and the downstream factors of the GGR pathway to the damaged strand. (B) Alignment of the RPA-B and XPC DNA-binding sequences. The consensus was derived using the following amino acid classes, in which lower case letters refer to the amino acid class and capital letters refer to the single letter amino acid code: hydrophobic (h, ALICVMYFW); the aliphatic subset of these (a, ALIVMC); small (s, ACDGNPSTV); the "tiny" subset of these (u, GAS); polar (p, CDEHKNQRST); charged (c, DEHKR), positively charged (+, HKR); and negatively charged (n, DE). The length of nonalignable gaps is indicated in parentheses and the beta-sheet elements are indicated by the arrows. [Reproduced from Maillard et al., (2007)]
An alternative model for damage recognition and NER has been developed by Reardon and Sancar (2005) to account for the efficiency of damage recognition by a suite of proteins that, individually, have weak affinity for damaged DNA (Figure 5). They suggest that the primary damage recognition proteins, XPC/TFIIH and XPA/RPA, interact with a damaged site, but require ATP hydrolysis by the XPB component of TFIIH to "lock" the proteins onto the damaged site (Reardon and Sancar, 2005). Irradiation of cells through micropores demonstrates the primary role for XPB in the transition (Oh et al., 2007). Cells with inactive XPB still recruit XPC and in some cases, XPA and XPG, but XPF is absent from the damaged sites (Oh et al., 2007).
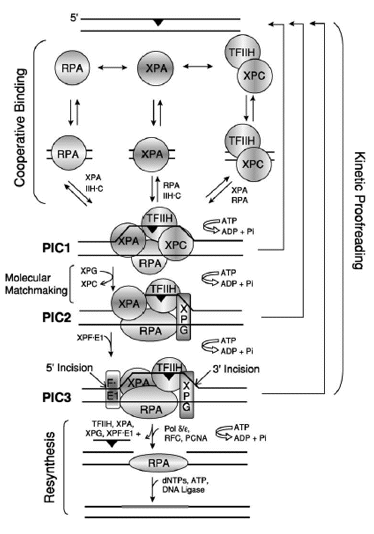
Figure. 5. Model for Excision Repair in Human Cells. The damage recognition factors, RPA, XPA, and XPC /TFIIH, assemble at the damage site (black triangles) in a random order but cooperative manner, to form an unstable ''closed'' complex. ATP hydrolysis by TFIIH unwinds the duplex around the lesion, causing the formation of a stable complex called preincision complex 1 (PIC1). XPC is a molecular matchmaker that helps recruit and deliver XPG to PIC1. XPC leaves prior to the formation of PIC2, which is composed of XPA, RPA, TFIIH, and XPG. Finally, this PIC2 complex is recognized by XPF/ERCC1, leading to the formation of PIC3 and the dual incision event, which releases damage in a 24-32 nucleotide-long oligomer. ATP hydrolysis is required for PIC1-PIC2-PIC3 formation, and the reaction may be aborted at any step along the pathway, leading to dual incision (kinetic proofreading). The excision gap is filled in by polymerase delta or epsilon (plus accessory factors), and the newly synthesized DNA (thick gray bar) is ligated to complete the repair reaction. The role played by DDB1/DDB2(XPE) in this process has not been characterized. [Reproduced from Reardon and Sancar, Nucleotide Excision Repair, in Progr Nucleic Acids Research and Mol Biol, 79, 183-235 2005, with permission of Elsevier]
Unwinding
The unwinding of DNA in the region around a damaged site requires components of the transcription factor TFIIH (Schaeffer et al., 1993). TFIIH is a general transcription factor for RNA Pol I and II, and the whole complex functions as a unit in repair (Figure 6). TFIIH has 10 component proteins of which XPB and XPD play a major role as unwinding helicases in NER. A subset of 6 proteins (XPB, XPD, p62, p52, p44, and p34) can be separated from the other components and form the core excision repair complex, which forms a ring structure around DNA (Schultz et al., 2000), and can support NER in vitro (Araujo et al., 2000). Different purification methods produce TFIIH that can copurify with XPC or XPG (Araujo et al., 2001).
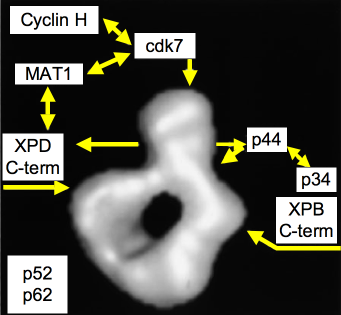
Figure 6. Model of the Quaternary Organization of TFIIH. The positions of subunits cdk7, XPD, XPB, and p44, as inferred from immunolabeling experiments, are indicated by arrows on the three-dimensional model of human TFIIH. Note that the anti-XPB and anti-XPD antibodies are directed against the extreme C terminus of the proteins. The N termini may interact directly in the lower part of the ring. The cyclin H, MAT1, and p34 repair are tentatively positioned on the model according to well-established subunit-subunit interaction studies. Subunits p62 and p52 have not been positioned. [Reproduced with modifications from Schultz et al. Molecular structure of human TFIIH. Cell, 102, 599-607, 2000 with permission of Elsevier]
XPB (ERCC3) (Weeda et al., 1990) and XPD (ERCC2) (Weber et al., 1990) are helicases of opposite polarity (3' to 5' and 5' to 3', respectively) that unwind the helix in the vicinity of the lesion. XPD contains a gated channel and an 4Fe-4S cluster that can sense damaged DNA, These 4Fe-4S cluster proteins become oxidized at the damaged sites, which enhances DNA binding. The other components of TFIIH do not have identified enzymatic activities but may function in the assembly and stabilization of TFIIH, and individual ones have specific interactions with XPB and XPD (Figure 6).
Mutations in XPB and XPD give rise to the complex human disorders of XP, CS and TTD, depending on the precise location of the mutation (see module on Human Photosensitive Diseases of DNA Repair). These mutations have been mapped onto the structure of XPD (Fan et al., 2008), and can be correlated with their cognate disease in three classes: (1) ATP and DNA binding for mutations causing XP, (2) conformation for mutations causing XP/CS, and (3) protein framework for mutations causing TTD.
XP mutations (Fan et al., 2008) are in the DNA- or ATP-binding channels, previously noted to be in helicase domains, and reduce DNA binding, helicase activity and bubble opening.
XP/CS mutations (Fan et al., 2008) can still bind DNA but cause the loss of helicase activity, and reduce the flexibility of HD1-HD2 domains of the protein. The altered conformational changes associated with ATP- and DNA-binding may affect protein-protein interactions within the TFIIH complex, especially with p44, and other critical protein partners including XPG. These conformationally restricted states have differential effects on transcription initiation versus NER or transcription-coupled repair. One consequence may be NER-dependent inappropriate incisions at transcription sites distant from DNA damage (Theron et al., 2005). XP/CS mutations can cause HD1-HD2 motifs to become locked in an abnormal conformation.
TTD mutations (Fan et al., 2008) cause framework defects that increase flexibility and impact the levels of XPD as well as the stability of TFIIH. Many of the XPD C-terminal modifications found in TTD weaken the interaction with p44, and disturb the conformation of TFIIH. Similarly, mutations in the p8 component affect the stability of TFIIH and are exclusively associated with TTD (Giglia-Mari et al., 2004).
Excision
Once the incision complexes have been positioned, the damaged site is excised by cleavages 3' and 5' to the damaged site (Figure 7). The footprint of XPA/RPA seems to define the sites for the 24-32 nucleotide fragment that is removed, with some variability according to the precise structure of the damage itself. The 3' cleavage is the second, requiring prior synthesis of the patch, by XPG (ERCC5), which is a 135-kDa protein that belongs to the FEN-1 family of structure-specific nucleases (Clarkson, 2003; Fagbemi et al., 2011). XPG preferentially binds to single strand-double strand junctions with strand specificity (Figure 7), and is involved in crosslink repair (Lee et al., 2000). XPG is downloaded by association with TFIIH, and interacts with RPA, and cleaves the junction only when the patch has been polymerized (Fagbemi et al., 2011).

Figure 7. Structure-Specific Activity of the 3' and 5' Nucleases on Opened DNA Structures. XPG makes the second 3' cleavage, and is downloaded to the damaged site by interactions with TFIIH and RPA. XPF/ERCC1 makes the initial 5' cleavage, and requires anchoring to XPA after the action of TFIIH and XPG.
The 5' cleavage is first, and is carried out by a heterodimer of two proteins, XPF and ERCC1, which are important for telomere structure (Zhu et al., 2003), crosslink repair (Zhang et al., 2000) and homologous recombination (Niedernhofer et al., 2004). Most nucleases attack double-strand breaks by resection of one strand in the 5' to 3' direction, leaving an overhang that resembles the substrate for XPF/ERCC1 in NER (Figure 7). Consequently, double-strand break repair that proceeds by single-strand annealing requires XPF/ERCC1. There is evidence that XPF/ERCC1 plays a different role in the NER of UV photoproducts, in comparison to its role in crosslink repair (Zhang et al., 2000). ERCC1 is a 33-kDa protein and XPF (ERCC4) is a 112-kDa protein; structural analysis suggests that the nuclease activity resides in the XPF partner. A metal-binding site is present in a nuclease motif, V/IERKX3D (Aravind et al., 1999), that is required for nuclease activity at the C terminus of XPF (Enzlin and Scharer, 2002). ERCC1 has strong interactions with XPA, and is essential for the enzymatic activity of XPF. Cells with mutant XPB, XPA and XPG can still download to damaged sites, but XPF/ERCC1 cannot (Oh et al., 2007).
Resynthesis
Once the oligonucleotide is removed, the resulting gap is filled in by the combined action of a DNA polymerase and ligase (Sancar, 1994). DNA repair in mammalian cells was first discovered though autoradiographic detection of the incorporation of [3H]-thymidine into repair patches after UV exposure (Figure 8) (Rasmussen and Painter, 1964, 1966). In proliferating and nonproliferating cells, the gap is filled in by Pol delta, PCNA, RPA and XRCC1/ligase III (Moser et al., 2007). The RPA may have remained following excision, because it was in place as an XPA/RPA heterodimer. In proliferating cells the gap is resynthesised by Pol epsilon and ligase I (Moser et al., 2007).
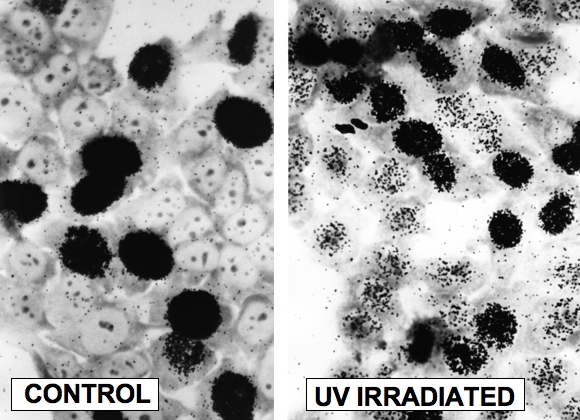
Figure 8. Autoradiographic Detection of DNA Repair (Unscheduled DNA Synthesis). Human fibroblasts were irradiated with approximately 20 Jm-2 UV (254 nm) light labeled with [3H]-thymidine and fixed 2 hr later. Cells were layered with Kodak NTB2 liquid photographic emulsion, and developed approximately 7 days later. Left: control cells showing intense labeling of S phase cells. Right: UV irradiated cells showing labeling of S phase cells, and lighter labeling of G1 and G2 cells carrying out NER (J.E. Cleaver, unpublished experiments; photography courtesy of G.H. Thomas).
Transcription Coupled Repair (TCR)
Early studies of DNA repair suggested that repair was not uniform in all parts of the genome. Repair of photoproducts in the alpha satellite sequence, a major repeat sequence, was found to be less efficient than in the rest of the genome (Zolan et al., 1982). The residual repair In XPC cells was shown to occur in domains, not randomly distributed, indicating that there was differential access to different parts of the genome that controlled the efficiency of repair (Mansbridge and Hanawalt, 1983). Subsequently these repaired regions in XPC cells were shown to support excision of photoproducts at the same rate as in wild-type cells (Karentz and Cleaver, 1986), and contain actively expressed genes (Venema et al., 1990a). Using sequence-specific probes, Mellon discovered that the highly expressed dhfr gene was more rapidly repaired than the remainder of the genome (Mellon et al., 1986), and that repair was faster in the transcribed strand than in the non-transcribed strand (Mellon et al., 1987). These observations that repair of actively transcribed genes was more rapid than the rest of the genome led to the definition of TCR as a distinct pathway of NER (Sancar, 1996; Wood, 1997; Hanawalt and Spivak, 2008).
The initiating signal for TCR is through the arrest of RNA Pol II at a damaged site, which is an alternative damage recognition mechanism to XPC/HR23B, which acts on non-transcribed DNA (Figure 2) (Ljungman and Zhang, 1996; Proietti De Santis et al., 2002; Svejstrup, 2002; D'Errico et al., 2003; Lindsey-Boltz and Sancar, 2007). This pathway is particularly important clinically, because deficiencies are associated with neurological disorders in XP and CS (Cleaver et al., 2009).
The arrest of Pol II may act as a potent apoptotic signal if not relieved by TCR (Ljungman and Zhang, 1996; Svejstrup, 2003). Arrested RNA Pol II is phosphorylated on its C-terminal domain, and subsequently poly-ubiquitinated (Bregman et al., 1996) to mark it for removal and degradation, leaving the active genes accessible for repair and the resumption of transcription (Yang et al., 2003). In E. coli, TCR is mediated by the Mfd protein; in mammalian cells the corresponding function is carried out by the CSA and CSB proteins, which are mutated in the human disease Cockayne syndrome (Venema et al. 1990b; Hanawalt and Spivak, 2008), and the UVSSA scaffold protein that is mutated in the human UVS disease (Cleaver, 2012).
CSA (ERCC8) is a WD40 protein that is a component of a ubiquitin ligase complex that ubiquitylates CSB targeting it for degradation releasing blocked forks (Henning et al., 1995; Shah and He, 2015). CSB (ERCC6) is SWI/SNF protein with ATPase activity that contains helicase motifs, but does not function as a helicase (Bohr, 1991; Licht et al., 2003). CSB facilitates the progress of RNA Pol II through damaged sites, and natural transcription pause sites (Bohr, 1991; Licht et al., 2003), and acts as a chromatin remodeling factor (Newman et al., 2006). In the absence of CSA or CSB, ubiquitination of the RNA Pol II C-terminal domain fails to occur (Bregman et al., 1996).
Early investigation of cells from CS patients showed that they failed to recover RNA and DNA synthesis after UV damage (Lehmann et al., 1979; Cleaver, 1982), and this property was used to demonstrate that there were two genes uniquely involved in the disease, and overlap with the other UV sensitive disease XP (Lehmann, 1982). Although the failure of RNA synthesis to recover after irradiation correlates with the reduced repair in actively transcribed genes, the failure of DNA synthesis has been more difficult to explain. Reduced DNA synthesis may be a passive result of replication blockage or could result from active signal transduction (Tresini et al., 2015). In E. coli, when the replication machinery collides head on with a transcription complex proceeding in the opposing direction, the RNA Pol II complex is displaced by the action of Mfd to allow DNA replication to resume (Pomerantz and O'Donnell, 2010). Mfd deficient cells have a persistent block to continued DNA replication. Although a similar function for CSA and CSB in displacing a blocked RNA Pol II complex could account for the failure of DNA synthesis to resume in human cells, the persistent DNA replication arrest involves the whole replicating genome, not just the actively expressed genes. The persistent delay in DNA replication may be the reason why CS cells do not show increased UV mutagenesis (Reid-Bayless et al., 2016) and CS patients do not develop skin cancer (Zhang et al., 2016).
TCR in Human Cancer
The capacity to sequence whole genomes has recently been applied to human cancers, producing considerable insight into the mechanism of carcinogenesis from chemicals or radiation. The genomic sequences of melanoma (Pleasance et al., 2010a) and lung cancer (Pleasance et al., 2010b) showed signature mutations corresponding to solar radiation or chemical exposure, respectively. Both tumors showed evidence for reduced mutagenesis due to TCR of the transcribed strands of expressed genes, and a more general expression-linked repair operating on both strands. The latter, expression-linked repair may correspond to the repaired regions originally identified in XPC cells that appeared to be considerably larger than individual genes (Mansbridge and Hanawalt, 1983; Karentz and Cleaver, 1986). Despite the action of TCR, many tumor specific driver mutations occur in actively expressed genes. The regional variations in mutation rates in cancers may be due to mismatch repair (Supek and Lehner, 2015), but transcription coupled repair of UV damage does not appear to play any role in mutagenesis or carcinogenesis (Reid-Bayless et al., 2016).
In skin cancers from XPC patients, most mutations arise from unrepaired damage in the nontranscribed strand of expressed genes (Zheng et al., 2014). The transcribed strand is repaired as efficiently as in tumors from non-XP patients, but this efficient repair confers no protection against the high rates of skin cancer in these patients (Zheng et al., 2014). In comparison CS patients have never been reported to develop skin cancer despite their strong photosensitivity (Zhang et al., 2106), and their cells do not show increased UV mutagenesis, despite the deficiency in TCR (Reid-Bayless et al., 2016).
DNA Replication on UV-Damaged Substrates
NER can remove DNA damage before DNA replication begins, and consequently plays a major role in reducing the amount of damage that becomes fixed as mutations during replication (Chen et al., 1990). Specialized polymerases are required to replicate DNA photoproducts, because the normal DNA polymerases, alpha, delta and epsilon cannot accommodate large distortions such as DNA photoproducts or adducts in their active sites (Brash et al., 1991; Steitz, 1999). There are 10 of these specialized polymerases, some of which have relaxed substrate specificity that can display error rates as high as 1% on undamaged DNA in vitro (Johnson et al., 2000b). The most important is the low fidelity polymerase, Pol eta (Johnson et al., 2000b, 1999; Masutani et al., 1999; Ohmori et al., 2001). This is mutated in the form of XP known as the XP variant, which is clinically similar to other XP patients, such as XP group C, both of which are without significant neurological complications (Broughton et al., 2002).
In the absence of Pol eta, bypass of UV photoproducts is achieved by its paralog Pol iota, but with increased error rates such that Pol iota is an actively mutagenic polymerase on UV damaged DNA (Gueranger et al., 2008). Pol zeta acts subsequent to Pol eta or Pol iota bypass, to complete the replication of a damaged site (Johnson et al., 2000a). Neither Pol eta nor Pol iota deficient cells show much increase in UV sensitivity, but the double mutant is very UV sensitive (Gueranger et al., 2008). A characteristic of cells lacking Pol eta is that their UV sensitivity is greatly increased by incubation in caffeine after UV exposure (Lehmann et al., 1975; Park and Cleaver, 1979). The most likely explanation for this response is that survival after UV irradiation in Pol eta deficient cells requires signal transduction during the S phase mediated by ATR which is sensitive to inhibition by caffeine (Sarkaria et al., 1999). Inhibition of ATR redirects cells into an ATM-dependent apoptotic pathway that resists inhibition by caffeine, despite the apparent sensitivity of ATM to caffeine in other assays (Sarkaria et al., 1999).
Chromatin Structure
Many histone modifications occur during normal DNA transactions as well as during damage responses, which appear conserved from yeast to human (Cosgrove et al., 2004; Downs and Cote, 2005; Garcia et al., 2007). These modifications act as binding sites for chromatin modifying complexes and transducing complexes, which mediate the damage response (Downs et al., 2004; Morrison et al., 2004; Murr et al., 2006; van Attikum et al., 2004). Early work showed that recently repaired sites were sensitive to degradation by microccal nuclease, suggesting that the chromatin structure was modified in the region of excision, and probably was less compact (Bodell et al., 1982; Cleaver, 1979).
Known histone modifications include acetylation, mono-, di-, and tri-methylation, phosphorylation, sumoylation, ubiquitination and ADP-ribosylation (Cosgrove et al., 2004; Garcia et al., 2007). These occur on both the histone tails that extend from the nucleosome core as well as on the core domains, and there can be 25 to 35 modifications on a single histone. The modifications to several histones are important in cell survival after DNA damage, including H2A, H2AX, H3 and H4 (Downs et al., 2004; Downs and Cote, 2005; Harvey et al., 2005). High mobility group protein B1 (HMGB1) is a multifunctional protein involved in chromatin structure, transcriptional regulation, V(D)J recombination, and inflammation. HMGB1 binds to and bends damaged DNA, and cells lacking HMGB1 show no histone acetylation upon DNA damage and reduced NER (Lange et al., 2008). The histone modification H3K9 is particularly important and high levels can reduce UV mutagenesis in human skin cancers (Zheng et al., 2014).
Phosphorylation of H2Ax (gamma-H2Ax), a minor histone, is an important cellular response to DNA breakage, arrested replication forks and other conformational changes (Lowndes and Toh, 2005), which activate the kinases ATM, ATR or DNA-dependent protein kinase. Gamma-H2Ax is visualized as discrete nuclear foci, which are related quantitatively to the frequency of DNA double-strand breaks, and is required for assembly of repair factors (Figure 9) (Lowndes and Toh, 2005; Park et al., 2003; Banath and Olive 2003; Rogakou et al., 1998, 1999; Wang et al., 2005; Ward et al., 2003;Paull et al., 2000; Rothkamm and Lobrich, 2003).
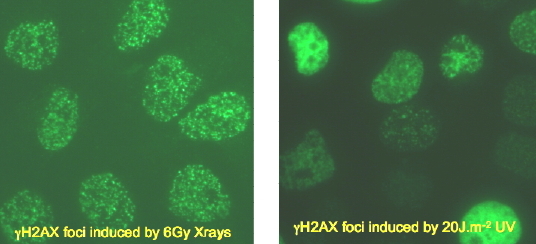
Figure 9. Induction of gamma-H2Ax in Response to Irradiation by X-rays or UV Radiation. Left: punctate foci of gamma-H2Ax formed at sites of DNA double-strand breaks. Right: complex distribution of gamma-H2Ax formed at sites of NER and replication arrest.
The role of gamma-H2Ax in response to UV-induced DNA damage is far less characterized, and gamma-H2Ax does not coalesce into similar quantifiable nuclear foci as it does on double-strand breaks (Figure 9) (Rothkamm and Lobrich, 2003). This is because negligible numbers of double-strand breaks are induced directly by UV radiation (see module on Double-Strand Breaks). The visual appearance of gamma-H2Ax fluorescence after UV irradiation consists of a range of foci, clustered patches and whole nuclear illumination (Figure 9) (Marti et al., 2006; Limoli et al., 2000, 2002; Halicka et al., 2005; Matsumoto et al., 2007). The latter may represent H2Ax displaced from chromatin, and is potentially an apoptotic signal (Liu, 2008).
Why Is Nucleotide Excision Repair So Complicated?
NER in eukaryotic cells employs the same principle as in prokaryotes: cleavage of the DNA on either side of the damaged site, and removal of the damaged oligonucleotide (Reardon and Sancar, 2005). Why then can E. coli complete the process with four genes (UvrA, B, C, D), whereas human cells need so many (Table 1)? Most human repair proteins function as members of heterodimers or more complex assemblages, adding to the total number of proteins involved.
A teleological view would be to say that the human genome is so complex that to find a few lesions in a large tangled mass of chromatin-compacted DNA requires a complicated search machinery. This "design" argument implies that chromatin came first, and repair systems later. The argument belies the fact that the eukaryotic genome and its maintenance machinery evolved in concert, and most likely arose by a fusion of several prokaryotic species, bacterial and archeal. This fusion of species cobbled together a mixture of proteins that sorted out a working compromise under evolutionary selection for cell survival, and other salient features that selection operated upon to lead to repair, as we know it. Interestingly, the efficiency of the human NER system is not maximized for the most common UV lesion, the CPD, but the rarer but more distorting [6-4]PD (Figure 1). Clearly a balance between the ease with which a distortion of the DNA helix can be recognized must be balanced against the distortion's frequency of occurrence, and its potential biological effectiveness.
The intermingling of excision repair components with other functions, especially transcription, replication and cell cycle regulation, is another striking feature of eukaryotic excision repair. In considering the evolution of excision repair, one possibility is that the primitive system was predominantly transcription coupled. This idea would be consistent with the suggestion that RNA polymerase is a more sensitive sensor of damage than XPC or XPE (Lindsey-Boltz and Sancar, 2007). Only with the development of multi-cellular organisms with somatic tissues would global genome repair have become important. The damage recognition proteins for global repair, XPC and XPE, could subsequently be recruited from ancestral single-strand binding proteins. Evolutionary optimizing of repair protein activity would then be balanced against optimizing other functions. The evolutionary trees of the human excision repair genes, especially those involved in damage recognition, might reveal potential common origins with other proteins involved in DNA-protein interactions.
References
Abraham, R.T. (2001) Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev, 15, 2177-2196.
Araujo, S.J., Nigg, E.A. and Wood, R.D. (2001) Strong functional interactions of TFIIH with XPC and XPG in human DNA nucleotide excision repair, without a preassembled repairosome. Mol Cell Biol, 21, 2281-2291.
Araujo, S.J., Tirode, F., Coin, F., Pospiech, H., Syvaoja, J.E., Stucki, M., Hubscher, U., Egly, J.M. and Wood, R.D. (2000) Nucleotide excision repair of DNA with recombinant human proteins: definition of the minimal set of factors, active forms of TFIIH, and modulation by CAK. Gene Devel, 14, 349-359.
Aravind, L., Walker, R.D. and Koonin, E.V. (1999) Conserved domains in DNA repair proteins and evolution of repair systems. Nucleic Acids Res, 27, 1223-1242.
Banath, J.P. and Olive, P.L. (2003) Expression of phosphorylated histone H2AX as a surrogate of cell likking by drugs that create DNA double-strand breaks. Cancer Res, 63, 4347-4350.
Barrett, S.F., J.H.Robbins, R.E.Tarone and K.H.Kraemer. (1991) Evidence for defective repair of cyclobutane dimers with normal repair of other photoproducts in a transcriptionally active gene transfected into Cockayne syndrome cells. Mutat Res, 255, 281-291.
Bodell, W.J., Kaufmann, W.K. and Cleaver, J.E. (1982) Enzyme digestion of intermediates of excision repair in human cells irradiated with ultraviolet light. Biochem, 21, 6767-6772.
Bohr, V.A. (1991) Gene specific DNA repair. Carcinogen, 12, 1983-1992.
Brash, D.E., J.A.Rudolph, J.A.Simon, A.Lin, G.J.McKenna, H.P.Baden, A.J.Halperin and J.Ponten. (1991) A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma. Proc. Natl. Acad. Sci. USA, 88, 10124-10128.
Bregman, D.B., Halaban, R., van Gool, A.J., Henning, K.A., Friedberg, E.C. and Warren.S.L. (1996) UV-induced ubiquitination of RNA polymerase II: a novel modification deficient in Cockayne syndrome cells. Proc. Natl. Acad. Sci. USA, 93, 11586-11590.
Broughton, B.C., Cordonnier, A., Kleijer, W.J., Jaspers, N.G., Fawcett, H., Raams, A., Garritsen, V.H., Stary, A., Avril, M.F., Boudsocq, F., Masutani, C., Hanaoka, F., Fuchs, R.P., Sarasin, A. and Lehmann, A.R. (2002) Molecular analysis of mutations in DNA polymerase eta in xeroderma pigmentosum-variant patients. Proc Natl Acad Sci USA., 99, 815-820.
Chen, R.H., Maher, V.M. and McCormick, J.J. (1990) Effect of excision repair by diploid human fibroblasts on the kinds and locations of mutations induced by (+/-)-7 beta,8 alpha-dihydroxy-9 alpha,10 alpha-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene in the coding region of the HPRT gene. Proc Natl Acad Sci USA., 87, 8680-8684.
Clarkson, S.G. (2003) The XPG story. Biochimie, 85, 1113-1121.
Cleaver, J.E. (1979) Similar distributions of repaired sites in chromatin of normal and xeroderma pigmentosum variant cells damaged by ultraviolet light. Biochim Biophys Acta, 565, 387-390.
Cleaver, J.E. (1982) Normal reconstruction of DNA supercoiling and chromatin structure in Cockayne syndrome cells during repair of damage from ultraviolet light. Amer J Hum Genet, 34, 566-575.
Cleaver, J.E. (2012) Photosensitivity syndrome brings to light a new transcription-coupled DNA repair cofactor. Nat Genet, 44, 477-478.
Cleaver, J.E., Lam,E.T., Revet, I. (2009) Disorders of nucleotide excision repair: the genetic and molecular basis of heterogeneity. Nat Rev Gen, 10, 756-768.
Cleaver, J.E. and Revet, I. (2008) Clinical implications of the basic defects in Cockayne syndrome and xeroderma pigmentosum and the DNA lesions responsible for cancer, neurodegeneration and aging. Mech Ageing Dev, 129, 492-497.
Cleaver, J.E. and Thomas, G.H. (1981) Measurement of unscheduled DNA synthesis by autoradiography. In Friedberg, E.C. and Hanawalt, P.C. (eds.), DNA repair: a laboratory manual of research procedures. Marcel Dekker Inc., New York and Basel, Vol. 1 Part B, pp. 277-287.
Cosgrove, M.S., Boeke, J.D. and Wolberger, C. (2004) Regulated nucleosome mobility and the histone code. Nat Struct Mol Biol, 11, 1037-1043.
D'Errico, M., Teson, M., Calcagnile, A., Proietti De Santis, L., Nikaido, O., Botta, E., Zambruno, G., Stefanini, M. and Dogliotti, E. (2003) Apoptosis and efficient repair of DNA damage protect human keratinocytes against UVB. Cell Death Differ, 10, 754-756.
Downs, J.A., Allard, S., Jobin-Robitaille, O., Javaheri, A., Auger, A., Bouchard, N., Kron, S.J., Jackson, S.P. and Cote, J. (2004) Binding of chromatin-modifying activities to phosphorylated histone H2A at DNA damage sites. Molec Cell, 16, 979-990.
Downs, J.A. and Cote, J. (2005) Dynamics of chromatin during the repair of DNA double-strand breaks. Cell Cycle, 4, 1373-1376.
Ellison, M.J. and Childs, J.D. (1981) Pyrimidine CPDs induced in Escherichia coli DNA by ultraviolet radiation present in sunlight. Photochem. Photobiol, 34, 465-469.
Emmert, S., Kobayashi, N., Khan, S.G. and Kraemer, K.H. (2000) The xeroderma pigmentosum group C gene leads to selective repair of cyclobutane pyrimidine dimers rather than 6-4 photoproducts. Proc Natl Acad Sci USA, 97, 2151-2156.
Enzlin, J.H. and Scharer, O.D. (2002) The active site of the DNA repair endonuclease XPF-ERCC1 forms a highly conserved nuclease motif. EMBO J, 21, 2045-2053.
Erixon, K. and Ahnstrom, G. (1979) Single-strand breaks in DNA during repair of UV-induced damage in normal human and xeroderma pigmentosum cells as determined by alkaline DNA unwinding and hydroxylapatite chromatography: effects of hydroxyurea, 5-fluorodeoxyuridine and 1-beta-D-arabinofuranosylcytosine on the kinetics of repair. Mutat Res, 59, 257-271.
Fagbemi A. F., Orelli, B., Scharer, O. D. (2011) Regulation of endonuclease activity in human nucleotide excision repair. DNA Repair (Amst), 10, 722-729.
Falck, J., Coates, J. and Jackson, S.P. (2005) Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature, 434, 605-611.
Fan, L., Fuss, J.O., Cheng, Q.J., Arvai, A.S., Hammel, M., Roberts, V.A., Cooper, P.K. and Tainer, J.A. (2008) XPD helicase structures and activities: insights into the cancer and aging phenotypes from XPD mutations. Cell, 133, 789-800.
Ford, J.M. (1998) Role of p53 in the mammalian cellular response to UV damage. Photochem Photobiol, 67, 73S-74S.
Ford, J.M., Lommel, L. and Hanawalt, P.C. (1994) Preferential repair of ultraviolet light-induced DNA damage in the transcribed strand of the human p53 gene. Mol Carcinogen, 10, 4734-4742.
Garcia, B.A., Shabanowitz, J. and Hunt, D.F. (2007) Characterization of histones and their post-translational modifications by mass spectrometry. Curr Opin Chem Biol, 11, 6-73.
Giglia-Mari, G., Coin, F., Ranish, J.A., Hoogstraten, D., Theil, A., Wijgers, N., Jaspers, N.G., Raams, A., Argentini, M., van der Spek, P.J., Botta, E., Stefanini, M., Egly, J.M., Aebersold, R., Hoeijmakers, J.H. and Vermeulen, W. (2004) A new, tenth subunit of TFIIH is responsible for the DNA repair syndrome trichothiodystrophy group A. Nat Genet, 36, 714-719.
Gueranger, Q., Stary, A., Aoufouchi, S., Faili, A., Sarasin, A., Reynaud, C.A. and Weill, J.C. (2008) Role of DNA polymerases eta, iota and zeta in UV resistance and UV-induced mutagenesis in a human cell line. DNA Repair, 7, 1551-1562.
Halicka, H.D., Huang, X., Traganos, F., King, M.A., Dai, W. and Darzynkiewicz, Z. (2005) Histone H2AX phosphorylation after cell irradiation with UV-B: relationship to cell cycle phase and induction of apoptosis. Cell Cycle, 4, 339-345.
Hanawalt, P.C., Spivak, G. (2008) Transcription-coupled DNA repair: two decades of progress and surprises. Nature Rev Mol Cell Biol, 9, 958-970.
Harvey, A.C., Jackson, S.P. and Downs, J.A. (2005) Saccharomyces cerevisiae histone H2A Ser122 facilitates DNA repair. Genetics, 170, 543-553.
Henning, K.A., Li, L., Iyer, N., McDaniel, D., Reagan, M.S., Legerski, R., Schultz, R.A., Stefanini, M., Lehmann, A.R., Mayne, L.V., Friedberg, E.C. (1995) The Cockayne syndrome group A gene encodes a WD repeat protein that interacts with the CSB protein and a subunit of RNA polymerase II, TFIIH. Cell, 82, 555-564.
Hoeijmakers, J.H. (2001) Genome maintenance mechanisms for preventing cancer. Nature, 411, 366-374.
Hurley, P.J. and Bunz, F. (2007) ATM and ATR components of an integrated circuit. Cell Cycle, 6, 414-417.
Itoh, T., Fujiwara, Y., Ono, T., Yamaizumi, .M. UVs syndrome, a new general category of photosensitive disorder with defective DNA repair, is distinct from xeroderma pigmentosum variant and rodent complementation group I. (1995) Am J Hum Genet, 56, 1267-1276.
Johnson, R.E., Kondratick, C.M., Prakash, S. and Prakash, L. (1999) hRAD30 mutations in the variant form of xeroderma pigmentosum. Science, 264, 263-265.
Johnson, R.E., Washington, M.T., Haracska, L., Prakash, S. and Prakash, L. (2000a) Eukaryotic polymerases i and z act sequentially to bypass DNA lesions. Nature, 406, 1015-1019.
Johnson, R.E., Washington, M.T., Prakash, S. and Prakash, L. (2000b) Fidelity of human DNA polymerase h. J Biol Chem, 275, 7447-7450.
Kantor, G.J., Barsalou, L.S. and Hanawalt, P.C. (1990) Selective repair of specific chromatin domains in UV-irradiated cells from xeroderma pigmentosum pigmentosum complementation group C. Mutat Res, 235, 171-180.
Karentz, D. and J.E.Cleaver. (1986) Excision repair in xeroderma pigmentosum group C but not group D is clustered in a small fraction of the total genome. Mutat Res, 165, 165-174.
Klimek, M. (1965) Formation but no excision of thymine dimers in mammalian cells after UV-irradiation. Neoplasma, 12, 459-460.
Lange, S.S., Mitchell, D.L. and Vasquez, K.M. (2008) High mobility group protein B1 enhances DNA repair and chromatin modification after DNA damage. Proc Natl Acad Sci USA, 105, 10320-10325.
Laposa, R.R., Feeney, L., Crowley, E., de Feraudy, S. and Cleaver, J.E. (2007) p53 suppression overwhelms DNA polymerase h deficiency in determining the cellular UV DNA damage response. DNA Repair, 6, 1794-1804.
Lee, Y.J., Park, S.J., Ciccone, S.L., Kim, C.R. and Lee, S.H. (2000) An in vivo analysis of MMC-induced DNA damage and its repair. Carcinogen, 27, 446-453.
Lehmann, A.R. (1982) Three complementation groups in Cockayne syndrome. Mutat Res, 106, 347-356.
Lehmann, A.R., Kirk-Bell, S., Arlett, C.A., Paterson, M.C., Lohman, P.H.M., de Weerd-Kastelein, E.A. and Bootsma, D. (1975) Xeroderma pigmentosum cells with normal levels of excision repair have a defect on DNA synthesis after UV-irradiation. Proc. Natl. Acad. Sci. USA, 72, 219-235.
Lehmann, A.R., Kirk-Bell, S., Mayne, L. (1979) Abnormal kinetics of DNA synthesis in ultraviolet light-irradiated cells from patients with Cockayne syndrome. Cancer Res, 39, 4237-4241.
Licht, C.L., Stevnser, T., Bohr, V.A. (2003) Cockayne syndrome group B cellular and biochemical functions. Amer J Human Genetics, 73, 1217-1239.
Limoli, C.L., Giedzinski, E., Bonner, W.M. and Cleaver, J.E. (2002) UV-induced replication arrest in the xeroderma pigmentosum variant leads to double strand breaks, g-H2Ax formation, and Mre11 relocalization. Proc Natl Acad Sci USA, 99, 233-238.
Limoli, C.L., Giedzinski, E., Morgan, W.F. and Cleaver, J.E. (2000) Polymerase h deficiency in the XP variant uncovers an overlap between the S phase checkpoint and double strand break repair. Proc Nat Acad Sci USA, 97, 7939-7946.
Lindsey-Boltz, L.A. and Sancar, A. (2007) RNA poymerase: the most specific damage recognition protein in cellular responses to DNA damage. Proc Nat Acad Sci USA, 104, 13213-13214.
Liu, Y., Parry, J.A., Chin, A., Duensing, S. and Duensing, A. (2008) Soluble histone H2AX is induced by DNA replication stress and sensitizes cells to undergo apoptosis. Mol Cancer, 7, 61-71.
Ljungman, M. and Zhang, F. (1996) Blockage of RNA polymerase as a possible trigger for u.v. light-induced apoptosis. Oncogene, 13, 823-831.
Lowndes, N.F. and Toh, G.W.-L. (2005) DNA repair: the importance of phosphorylating histone H2AX. Curr Biol, 15, R99-R102.
Maillard, O., Solyom, S. and Naegeli, H. (2007) An aromatic sensor with aversion to damaged strands confers versatility to DNA repair. PloS Biology, 5, e79.
Mansbridge, J.N. and Hanawalt, P.C. (1983) Domain-limited repair of DNA in ultraviolet irradiated fibroblasts from xeroderma pigmentosum complementaiton C. In Friedberg, E.C. and Bridges, B.R. (eds.), Cellular responses to DNA damage. UCLA Symposium on molecular and cellular biology,New series. Alan R.Liss, New York, Vol. II, pp. 195-207.
Marti, T.M., Hefner, E., Feeney, L., Natale, V. and Cleaver, J.E. (2006) H2AX phosphorylation within the G1 phase after UV irradiation depends on nucleotide excision repair and not DNA double strand breaks. Proc Natl Acad Sci USA, 103, 9891-9896.
Masutani, C., Kusumoto, R., Yamada, A., Dohmae, N., Yokol, M., Yuasa, M., Araki, M., Iwa, S., Takio, K. and Hanoaka, F. (1999) The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase h. Nature, 399, 700-704.
Matsumoto, M., Yaginuma, K., Igarashi, A., Imura, M., Hasegawa, M., Iwabuchi, K., Date, T., Mori, T., Ishizaki, K., Yamashita, K., Inobe, M. and Matsunaga, T. (2007) Perturbed gap-filling synthesis in nucleotide excision repair causes histone H2AX phosphorylation in human quiescent cells. J Cell Sci, 120, 1104-1112.
Matsuoka, S., Ballif, B.A., Smogorzewska, A., McDonald, E.R.r., Hurov, K.E., Luo, J., Bakalarski, C.E., Zhao, Z., Solimini, N., Lerenthal, Y., Shiloh, Y., Gygi, S.P. and Elledge, S.J. (2007) ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science, 316, 1160-1166.
Mellon, I., Bohr, V.M., Hanawalt, P.C. (1986) Preferential repair of an active gene in human cells. Proc Nat Acad Sci USA, 83, 8878-8882.
Mellon, I., Spivak, G., Hanawalt, P.C. (1987) Selective removal of transcription-blocking DNA damage from the transcribed strand of the mammalian DHFR gene. Cell, 51, 241-249.
Mitchell, D.L., C.A.Haipe and J.M.Clarkson. (1985) (6-4) photoproducts are removed from the DNA of UV-irradiated mammalian cells more efficiently than cyclobutane dimers. Mutat Res, 143, 109-112.
Mitchell, D.L. and Cleaver, J.E. (1990) Photochemical alterations of cytosine account for most biological effects after ultraviolet irradiation. Trends in Photochem and Photobiol, 1, 107-119.
Mitchell, D.L., Greinert, R., de Gruijl, F.R., Guikers, K.L., Breitbart, E.W., Byrom, M., Gallmeier, M.M., Lowery, M.G. and Volkmer, B. (1999) Effects of chronic low-dose ultraviolet B radiation on DNA damage and repair in mouse skin. Cancer Res, 59, 2875-2884.
Mitchell, D.L., J.E.Cleaver and J.H.Epstein. (1990a) Repair of pyrimidine (6-4) pyrimidinone photoproducts in mouse skin. J. Inves Derm, 95, 55-59.
Mitchell, D.L., Nguyen, T.D. and Cleaver, J.E. (1990b) Nonrandom induction of pyrimidine-pyrimidone [6-4] photoproducts in ultraviolet-irradiated human chromatin. J. Biol. Chem, 265, 5353-5356.
Moll, P.R., Sander, V., Frischauf, A.M. and Richter, K. (2007) Expression profiling of vitamin D treated primary human keratinocytes. J Cell Biochem, 100, 574-592.
Morrison, A.J., Highland, J., Krogan, N.J., Arbel-Eden, A., Greenblatt, J.F., Haber, J.E. and Shen, X. (2004) INO80 and gH2AX interaction links ATP-dependent chromatin remodeling to DNA damage repair. Cell, 119, 767-775.
Moser, J., Kool, H., Giakzidis, I., Caldecott, K., Mullenders, L.H. and Fousteri, M.I. (2007) Sealing of chromosomal DNA nicks during nucleotide excision repair requires XRCC1 and DNA ligase III alpha in a cell-cycle-specific manner. Mol Cell, 27, 311-323.
Murr, R., Loizou, J.I., Yang, Y.G., Cuenin, C., Li, H., Wang, Z.Q. and Herceg, Z. (2006) Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat Cell Biol, 8, 91-99.
Newman, J.C., Bailey, A.D. and Weiner, A.M. (2006) Cockayne syndrome group B protein (CSB) plays a general role in chromatin maintenance and remodeling. Proc Nat Acad Sci USA, 103, 9613-9618.
Niedernhofer, L.J., Odijk, H., Budzowska, M., van Drunen, E., Maas, A., Theil, A.F., de Wit, J., Jaspers, N.G., Beverloo, H.B., Hoeijmakers, J.H. and Kanaar, R. (2004) The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks. Mol Cell Biol, 24, 5776-5787.
Oh, K.S., Imoto, K., Boyle, J., Khan, S.G. and Kraemer, K.H. (2007) Influence of XPB helicase on recruitment and redistribution of nucleotide excision repair proteins at sites of UV-induced DNA damage. DNA Repair (Amst), 6, 1359-1370.
Ohmori, H., Friedberg, E.C., Fuchs, R.P.P., Goodman, M.F., Hanaoka, F., Hinkle, D., Kunkel, T.A., Lawrence, C.W., Livneh, Z., Nohmi, T., Prakash, L., Prakash, S., Todo, T., Walker, G.C., Wang, Z. and Woodgate, R. (2001) The Y-Family of DNA Polymerases. Molecular Cell, 8, 7-8.
Painter, R.B. and Cleaver, J.E. (1969) Repair replication, unscheduled DNA synthesis, and the repair of mammalian DNA. Rad Res, 37, 451-466.
Park, E.J., Chan, D.W., Park, J.H., Oettinger, M.A. and Kwon, J. (2003) DNA-PK is activated by nucleosomes and phosphorylates H2AX within the nucleosomes in an acetylation-dependent manner. Nucleic Acids Res, 31, 6819-6827.
Park, S.D. and J.E.Cleaver. (1979) Postreplication repair: questions of its definition and possible alterations in xeroderma pigmentosum cell strains. Proc. Natl. Acad. Sci. USA, 76, 3927-3931.
Parris, C.H. and Kraemer, K.H. (1993) Ultraviolet-light induced mutations in Cockayne syndrome cells are primarily caused by cyclobutane dimer photoproducts while repair of other photoproducts is normal. Proc. Natl. Acad. Sci. USA, 90, 7260-7264.
Paull, T.T., Rogakou, E.P., Yamazaki, V., Kirchgessner, C.U., Gellert, M. and Bonner, W.M. (2000) A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol, 10, 886-895.
Pleasance, E.D., Cheetham, R.K., Stephens, P.J., McBride, D.J., Humphray, S.J., Greenman, C.D., Varela, I., Lin, M.L., Ordonez, G.R., Bignell, G.R., Ye, K., et al. (2010a) A comprehensive catalogue of somatic mutations from a human cancer genome. Nature, 463, 191-196.
Pleasance, E.D., Stephens, P., O'Meara, S., McBride, D.J., Meynert, A., Jones, D., Lin, M.L., Beare, D., Lau, K.W., Greenman, C., Varela, I., et al. (2010b) A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature, 463, 184-190.
Pomerantz, R.T., O'Donnell, M. (2010) Direct restart of a replication fork stalled by a head-on RNA polymerase. Science, 327, 590-592.
Proietti De Santis, L., Garcia, C.L., Balajee, A.S., Latini, P., Pichierri, P., Nikaido, O., Stefanini, M. and Palitti, F. (2002) Transcription coupled repair efficiency determines the cell cycle progression and apoptosis after UV exposure in hamster cells. DNA Repair, 1, 209-223.
Rasmussen, R.E. and Painter, R.B. (1964) Evidence for repair of ultraviolet damaged deoxyribonucleic acid in cultured mammalian cells. Nature, 203, 1360-1362.
Rasmussen, R.E. and Painter, R.B. (1966) Radiation-stimulated DNA synthesis in cultured mammalian cells. J Cell Biol, 29, 11-19.
Reardon, J.T., Nichols, A.F., Keeney, S., Smith, C.A., Taylor, J.S., Linn, S. and Sancar, A. (1993) Comparative analysis of binding of human damaged DNA-binding protein (XPE) and Escherichia coli damage recognition binding protein (UvrA) to the major ultraviolet photoproducts: T[c,s]T, T[t,s]T, T[6-4]T, and T[Dewar]T. J. Biol. Chem., 268, 21301-21308.
Reardon, J.T. and Sancar, A. (2005) nucleotide excision repair. Progr nucleic acids research and mol biol, 79, 183-235.
Reid-Bayless, K., Loeb, L., Aaron, S.T., Bezrookove, V., Cleaver, J.E. (2016) Why Cockayne syndrome patients do not get cancer despite their DNA repair deficiency. Proc Natl. Acad Sci USA (in press).
Robu, M., Shah,R.G., Petitclerc, N., Brind'Amour,J., Kandan-Kulangara,F., Shah.G.M. (2013) Role of poly(ADP-ribose) polymerase-1 in the removal of UV-induced DNA lesions by nucleotide excision repair. Proc. Natl. Acad. Sci. USA. 110:1658-1663.
Rogakou, E.P., Boon, C., Redon, C. and Bonner, W.M. (1999) Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol, 146, 905-916.
Rogakou, E.P., Pilch, D.R., Orr, A.H., Ivanova, V.S. and Bonner, W.M. (1998) DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem., 273, 5858-5868.
Rothkamm, K. and Lobrich, M. (2003) Evidence for a lack of DNA double strand break repair in human cells exposed to very low X-ray doses. Proc. Natl.Acad. Sci USA, 100, 5057-5062.
Sancar, A. (1994) Mechanisms of DNA excision repair. Science, 266, 1954-1956.
Sancar, A. (1996) DNA excision repair. Ann. Rev. Biochem, 65, 43-81.
Sarkaria, J.N., Busby, E.C., Tibbetts, R.S., Roos, P., Taya, Y., Karnitz, L.M. and Abraham, R.T. (1999) Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res, 59, 4375-4382.
Schaeffer, L., Roy, R., Humbert, S., Moncollin, V., Vermeulen, W., Hoeijmakers, J.H.J., Chambon, P. and Egly, J.M. (1993) DNA repair helicase: a component of BTF2 (TFIIH) basic transcription factor. Science, 260, 58-63.
Scheibye-Knudsen M, Croteau DL, Bohr VA. (2013) Mitochondrial deficiency in Cockayne syndrome. Mech Ageing Dev, (2013) 134(5-6):275-283.
Schultz, P., Fribourg, S., Poterszman, A., Mallouh, V., Moras, D. and Egly, J.M. (2000) Molecular structure of human TFIIH. Cell, 102, 599-607.
Shah, P., He, Y-Y. (2015) Molecular regulation of UV-induced repair. Photochem Photobiol, 91, 254-264.
Steitz, T.A. (1999) DNA polymerases: structural diversity and common mechanisms. J. Biol. Chem., 274, 17395-17398.
Supek,F., Lehner,B. 2015, Differential DNA mismatch repair underlies mutation rate variation across the human genome. Nature,521:81-84.
Sugasawa, K., Okuda, Y., Saijo, M., Nishi, R., Matsuda, N., Chu, G., Mori, T., Iwai, S., Tanaka, K., Tanaka, K. and Hanaoka, F. (2005) UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell, 121, 387-400.
Svejstrup, J.Q. (2002) Mechanisms of transcription-coupled DNA repair. Nat Rev Mol Cell Biol, 3, 21-29.
Svejstrup, J.Q. (2003) Rescue of arrested RNA polymerase II complexes. J Cell Sci, 116(Pt 3), 447-451.
Tang, J. and Chu, G. (2002) Xeroderma pigmentosum complementation group E and UV-damaged DNA-binding protein. DNA Repair, 1, 601-616.
Tang, J.Y., Hwang, B.J., Ford, J.M., Hanawalt, P.C. and Chu, G. (2000) Xeroderma pigmentosum p48 gene enhances global genomic repair and suppresses UV-induced mutagenesis. Molecular Cell, 5, 737-744.
Tang, M.-S., Hrncir, J., Mitchell, D., Ross, J. and Clarkson, J. (1986) The relative cytotoxicity and mutagenicity of cyclobutane pyrimidine dimers and [6-4] photoproducts in Escherichia coli cells. Mutat Res, 161, 9-17.
Taylor, J.S. and Cohrs, M.P. (1987) DNA, light. and Dewar pyrimidinones: the structure and significance of TpT3. J Amer Chem Soc, 109, 2834-2835.
Theron, T., Fousteri, M.I., Volker, M., Harries, L.W., Botta, E., Stefanini, M., Fujimoto, M., Andressoo, J.O., Mitchell, J., Jaspers, N.G., McDaniel, L.D., Mullenders, L. and Lehmann, A.R. (2005) Transcription-associated breaks in xeroderma pigmentosum group D cells from patients with combined features of xeroderma pigmentosum and Cockayne syndrome. Mol Cell Biol, 25, 8368-8378.
Tresini, M., Warmerdam, D. O., Kolovos, P., Snijder, L., Vrouwe, M.G., Demmers, J.A., van IJcken, W.F., Grosveld, F.G., Medema, R.H., Hoeijmakers, J.H., Mullenders, L.H., Vermeulen, W., Marteijn, J.A. (2015) The core spliceosome as target and effector of non-canonical ATM signalling. Nature, 523, 53-58.
van Attikum, H., Fritsch, O., Hohn, B. and Gasser, S.M. (2004) Recruitment of the INO80 complex by H2A phosphorylation links ATP-dependent chromatin remodeling with DNA double-strand break repair. Cell, 119, 777-788.
Venema, J., Hoffen, A.V., Natarajan, A.T., van Zeeland, A.A., Mullenders, L.H. (1990a) The residual repair capacity of xeroderma pigmentosum group C fibroblasts is highly specific for transcriptionally active DNA. Nuc Acids Res,18, 443-448.
Venema, J., Mullenders, L.H., Natarajan, A.T., van Zeeland, A.A., Mayne, L.Y. (1990b) The genetic defect in Cockyne syndrome is associated with a defect in repair of UV-induced DNA damage in transcriptionally active DNA. Proc Nat Acad Sci USA, 87, 4707-4711.
Volker, M., Mone, M.J., Karmakar, P., van Hoffen, A., Schul, W., Vermeulen, W., Hoeijmakers, J.H., van Driel, R., van Zeeland, A.A. and Mullenders, L.H. (2001) Sequential assembly of the nucleotide excision repair factors in vivo. Molec Cell, 8, 213-224.
Wang, H., Wang, M., Wang, H., Bocker, W. and Iliakis, G. (2005) Complex H2AX phosphorylation patterns by multiple kinases including ATM and DNA-PK in human cells exposed to ionizing radiation and exposed to kinase inhibitors. J Cell Physiol, 202, 492-502.
Ward, I.M. and Chen, J. (2001) Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem, 276, 47759-47762.
Ward, I.M., Minn, K., Jordan, K.J. and Chen, J. (2003) Accumulation of checkpoint protein 53BP1 at DNA breaks involves its binding to phosphorylated histone H2AX. TJ Biol Chem, 278, 19579-19582.
Weber, C.A., E.P.Salazar, S.A.Stewart and L.H.Thompson. (1990) ERCC2: cDNA cloning and molecular characterization of a human nucleotide excision repair gene with high homology to yeast RAD3. EMBO Journal, 9, 1437-1447.
Weeda, G., Ham, R.C.A.v., Vermeulen, W., Bootsma, D., Eb, A.J.v.d. and Hoeijmakers, J.H.J. (1990) A presumed helicase encoded by ERCC-3 is involved in the human repair disorders xeroderma pigmentosum and Cockayne's syndrome. Cell, 62, 777-791.
Wood, R.D. (1997) Nucleotide excision repair in mammalian cells. J Biol Chem, 272, 23465-23468.
Wood, R.D. (1999) DNA damage recognition during nucleotide excision repair in mammalian cells. Biochimie, 81, 39-44.
Wood, R.D. and Lindahl, T. (1999) Quality control by DNA repair. Science, 286, 1897-1905.
Wood, R.D., Mitchell,M., Sgouros,J., Lindahl,T. (2001) Human DNA repair genes. Science, 291, 1284-1289.
Yang, L.J., Jiang, H. and Rangel, K.M. (2003) RNA polymerase II stalled on a DNA template during transcription elongation is ubiquitinated and the ubiquitination facilitates displacement of the elongation complex. Int J Oncol, 22, 683-689.
Zelle, B. and Lohman, P.H. (1979) Repair of UV-endonuclease-susceptible sites in the 7 complementation groups of xeroderma pigmentosum A through G. Mutat Res, 62, 363-368.
Zhang, E.T, He, Y., Grob, P., Fong, Y.W., Nogales, E., Tjian, R. (2015) Architecture of the human XPC DNA repair and stem cell coactivator complex. Proc. Natl. Acad. Sci. USA, 112,14817-14822.
Zhang, W.R., Garrett, G.L., Cleaver, J.E., Arron, S.T. (2016) Absence of skin cancer in the DNA repair-deficient disease Cockayne Syndrome (CS): A survey study. J Am Acad Dermatol, 74:1270-1272.
Zhang, N., Zhang, X., Peterson, C., Li, L. and Legerski, R. (2000) Differential processing of UV mimetic and interstrand crosslink damage by XPF cell extracts. Nucleic Acids Res, 28, 4800-4804.
Zheng, C.L., Wang, N.J., Chung, J., Moslehi, H., Sanborn, J.Z,, Hur, J.S., Collisson, E.A., Vemula, S.S., Naujokas, A., Chiotti, K.E., Cheng, J.B., Fassihi, H., Blumberg, A.J., Bailey, C.V., Fudem, G.M., Mihm, F.G., Cunningham, B.B., Neuhaus, I.M., Liao, W., Oh, D.H., Cleaver, J.E., LeBoit, P.E., Costello, J.F., Lehmann, A.R., Gray, J.W., Spellman, P.T., Arron, S.T., Huh, N., Purdom, E., Cho, R.J. (2014) Transcription restores DNA repair to heterochromatin, determining regional mutation rates in cancer genomes. Cell Rep, 20,1228-1234.
Zhu, X.D., Niedernhofer, L., Kuster, B., Mann, M., Hoeijmakers, J.H. and de Lange, T. (2003) ERCC1/XPF removes the 3' overhang from uncapped telomeres and represses formation of telomeric DNA-containing double minute chromosomes. Mol Cell, 12, 1498-1498.
Zolan, M.E., Cortopassi, G.A., Smith, C.A., Hanawalt, P.C. (1982) Deficient repair of chemical adducts in alpha DNA of monkey cells. Cell, 28, 613-619.
11/08/08
03/04/10
09/01/16