LIGHT and CHLOROPLAST ENZYME ACTIVITY
Peter Schürmann
Université de Neuchâtel, Laboratoire de biologie moléculaire et cellulaire
Rue Emile-Argand 11, CP 158, CH-2009 Neuchatel, Switzerland
peter.schurmann@unine.ch
Introduction
Light provides the energy needed for the assimilation of carbon by photosynthesis. Light energy absorbed by the photosystems is converted into chemical energy in the form of ATP and NADPH, which serve as co-substrates in the reduction of CO2 to the level of carbohydrates by enzyme reactions of the reductive pentose phosphate cycle or Calvin-Benson cycle. However, this is not the only task light accomplishes in photosynthesis. Light functions also as an essential regulatory element, controlling the activity of enzymes involved in carbon assimilation and related metabolic pathways in chloroplasts. Light activates photosynthetic, anabolic pathways and, at the same time, deactivates catabolic processes, which are required for dark metabolism in the chloroplast. This dual control prevents the operation of futile cycles, which would occur if these opposing reaction pathways were allowed to function simultaneously. In addition, as light is variable in terms of quantity and quality, plants, particularly vascular plants that live at fixed locations, have evolved intricate regulatory networks to adjust their metabolism to changing environmental conditions in order to optimize harnessing light. In this module, I will describe light-dependent mechanisms that regulate the activity of key enzymes in photosynthetic metabolism, and discuss in more detail the ferredoxin/thioredoxin system, which is unique to oxygenic photosynthetic cells. Several recent reviews have addressed different aspects of the regulation of photosynthetic enzymes and provide further detailed information on the topic (Buchanan & Balmer, 2005; Gontero et al., 2006; Mora-Garcia et al., 2006; Schürmann & Buchanan, 2008; Meyer et al., 2009). Instead of citing many original articles I have preferred to refer to reviews wherein the pertinent references can be found.
Light-Induced Changes in Chloroplasts
Early experiments with algal cells suggested that light acts as a regulatory signal by activating certain carbon assimilation enzymes, enabling the cells to switch between dark and light metabolism. Different mechanisms have since been found to be engaged in this light dependent regulation, all based on changes of ambient conditions in the chloroplast stroma. Parameters such as pH, concentration of Mg2+ and metabolites, and redox potential are subject to light-induced alterations that influence the activity of susceptible enzymes (Buchanan, 1980).
Coupled to the light-driven electron transport between the photosystems, protons are translocated from the stroma across the thylakoid membrane into the thylakoid lumen (Figure 1). This increases the stromal pH from about pH 7 in the dark, to pH 8 in the light. Such an alkalization by one pH unit affects all stromal enzymes whose activity is strongly pH dependent, and thus serves as a general regulatory factor by providing a favorable pH for activity.
The transport of protons into the thylakoid lumen is accompanied by a release of Mg2+, the major counter-ion, from the thylakoid membrane into the stroma. This brings about a 1-3 mM increase of the stromal Mg2+ concentration, stimulating the activity of several enzymes that depend on Mg2+ for optimal function. The change in the Mg-ion concentration, therefore, represents another general regulatory element capable of modifying enzyme activities in response to light.
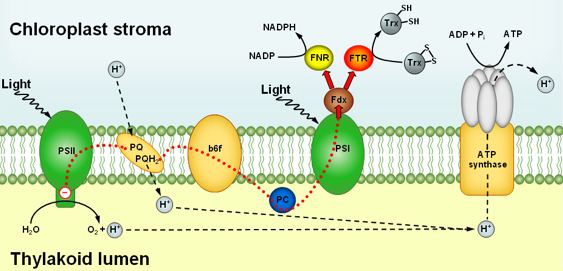
Figure 1: Light-dependent electron and proton transport in the thylakoid membrane and associated reactions. Electrons released in the oxidation of H2O are transferred in the light through photosystem I (PSI), plastoquinone (PQ), the cytochrome b6f complex, plastocyanin (PC), and photosystem II to ferredoxin (Fdx). Reduced Fdx reduces either thioredoxins (Trx) via ferredoxin:thioredoxin reductase (FTR) or NADP via ferredoxin:NADP reductase (FNR). Protons released into the thylakoid lumen in the oxidation of H2O and by PQ are used for the synthesis of ATP (Figure modified from Meyer et al., 2009).
A third important parameter undergoing light-dependent changes is redox potential. In the light, the stroma becomes much more reducing due to electrons delivered to ferredoxin by non-cyclic electron transport (Figure 1). This redox change is sensed and transmitted to selected enzymes by a redox cascade known as the ferredoxin/thioredoxin system. It is composed of three proteins, ferredoxin (Fdx), ferredoxin:thioredoxin reductase (FTR), and thioredoxin (Trx), and causes reversible structural changes in the target proteins that modify their catalytic activity. This regulatory system is more specific than pH and Mg2+ ion concentration changes, since it entails specific protein-protein interactions within the redox cascade, and with selected target enzymes. Consequently, only those proteins that have developed a redox sensitive "receiver" structure are affected.
Components of the Ferredoxin/Thioredoxin System
All components of the regulatory cascade are small, soluble stromal proteins containing either an iron-sulfur cluster, a redox-active disulfide bridge or both. They have been extensively studied, and their structures determined (Dai et al., 2000).
The first protein in the chain, receiving electrons from photosystem 1 (Figure 1), is ferredoxin. Plant-type Fdxs are 11 kDa acidic proteins that contain a single [2Fe-2S] cluster with a low redox potential of approximately -470 mV at pH 7.9, the pH of the stroma upon illumination. Fdx can carry one electron and deliver it to FTR in a non-covalent interaction between the two proteins. It is the first soluble electron carrier in the chloroplast, and distributes electrons to a number of enzymes in addition to FTR. The most important is Fdx:NADP reductase, the flavoprotein that produces NADPH needed for the reductive step in the Calvin-Benson cycle. Enzymes involved in N- and S-metabolism, i.e., nitrite reductase, glutamate synthase, and sulfite reductase, also obtain electrons from Fdx (Hase et al., 2006).
Ferredoxin:thioredoxin reductase, exclusively found in oxygenic photosynthetic cells, is the central enzyme of the regulatory cascade. It is an
 ß-heterodimer composed of a variable (7-13 kDa) and a catalytic (13 kDa) subunit. Size and primary structure of the variable subunit vary significantly from different species, whereas the catalytic subunit is highly conserved, including the arrangement of 6 Cys in two Cys-Pro-Cys, and one Cys-His-Cys motifs. This latter subunit contains the essential elements for catalysis, a [4Fe-4S] cluster and an adjacent redox-active disulfide. FTR is a flat, concave, disk-like molecule, well adapted to its function that necessitates simultaneous interaction with electron donor and acceptor (Figure 2).
ß-heterodimer composed of a variable (7-13 kDa) and a catalytic (13 kDa) subunit. Size and primary structure of the variable subunit vary significantly from different species, whereas the catalytic subunit is highly conserved, including the arrangement of 6 Cys in two Cys-Pro-Cys, and one Cys-His-Cys motifs. This latter subunit contains the essential elements for catalysis, a [4Fe-4S] cluster and an adjacent redox-active disulfide. FTR is a flat, concave, disk-like molecule, well adapted to its function that necessitates simultaneous interaction with electron donor and acceptor (Figure 2).
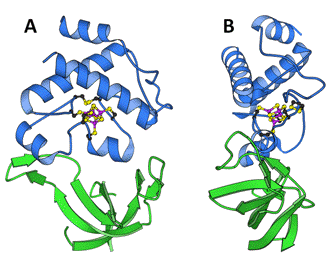
Figure 2. Crystal structure of the FTR heterodimer seen from front (A) and from the side (B). The variable subunit (green) is a heart-shaped, open ß-barrel structure, and the catalytic subunit (blue) an entirely
The [4Fe-4S] cluster is close to the surface that carries some positive charges on one side. The redox-active disulfide is on the opposite side whose surface is more hydrophobic, thus permitting interaction with different types of Trxs. Both FTR surfaces are highly conserved, allowing simultaneous docking of negatively charged Fdx on the cluster side and Trx on the disulfide side (Figure 3) (Dai et al., 2007; Xu et al., 2009).

Figure 3. Crystal structure of the ternary complex Fdx-FTR-Trx f. Whereas the interaction between Fdx (blue) and FTR (beige, brown) is noncovalent, Trx f (green) is covalently linked through a disulfide bond (colored in yellow) between the active sites of the two proteins (Figure taken from Dai et al., 2007). This complex represents the transient one-electron-reduced reaction intermediate, which has been stabilized by using an active-site mutant of Trx f (Glauser et al., 2004).
The third member of the regulatory cascade is thioredoxin. Trxs are ubiquitous, low-molecular weight protein disulfide reductases with a large number of biological functions (Arner & Holmgren, 2000). They catalyze thiol-disulfide exchange reactions based on reversible oxidation of two cysteine thiol groups to a disulfide, accompanied by the transfer of two protons. Plants contain a large number of Trxs varying in primary structure, function and location (Meyer et al., 2008). Four different types are found in chloroplasts, Trx f, m, x and y, which all share the canonical active-site sequence -Trp-Cys-Gly-Pro-Cys-. However, only Trxs f and m are involved in enzyme regulation; the other two appear to function in detoxification (removal of reactive oxygen species, ROS). Trx f, originally described as a specific activator protein for chloroplast fructose-1,6-bisphosphatase, is of eukaryotic origin and present only in this group of organisms. Discovered as an activator protein for chloroplast NADP-dependent malate dehydrogenase in C3 and C4 plants Trx m is found in oxygenic prokaryotes, algae and land plants. All four chloroplast Trxs are reduced by FTR in the light, and reoxidized by O2 or other oxidants in the dark. The chloroplast Trxs have the typical basic structure observed in all known Trxs (Figure 4).
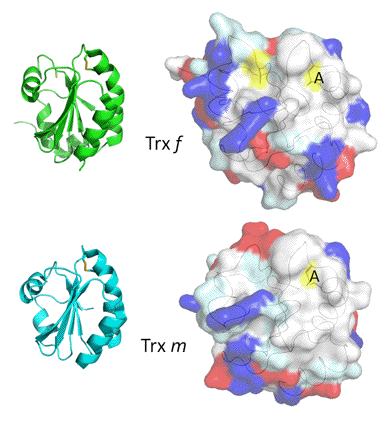
Figure 4. Crystal structures of spinach Trx f and Trx m. The two Trxs have the same overall 3-D structure (cartoon representations to the left), with a central five-stranded ß-sheet, surrounded by four
The surface properties are probably responsible for specificity in the protein-protein interactions. Several targets are very selective, and are activated efficiently only by Trx f, whereas others are also activated by Trx m. Since experiments exploring Trx specificity have not always been performed under the same conditions or with proteins from the same species, certain results differ. In general, results favor Trx f as the primary activator protein. So far, a target specific for Trx m has not been identified.
The Mechanism of Reduction of Target Enzymes by Thioredoxin
On its way from the thylakoid membranes to target enzymes, the light signal is transformed from an electron signal to a dithiol signal that can be perceived by proteins possessing a regulatory disulfide bridge. This transformation is performed by FTR through a novel mechanism involving a [4Fe-4S] cluster, and an associated disulfide bridge (Walters et al., 2005, 2009). The proposed mechanism accommodates defined features of the participants: (i) the reduction of a disulfide bond requires two electrons; (ii) Fdx is a one-electron donor; and (iii) FTR has a single Fdx docking site. Hence, the two-step mechanism proposed involves two consecutive electron transfers from Fdx to FTR, with the concomitant formation of a transient one-electron-reduced intermediate stabilized by the [4Fe-4S] cluster (Figure 5).
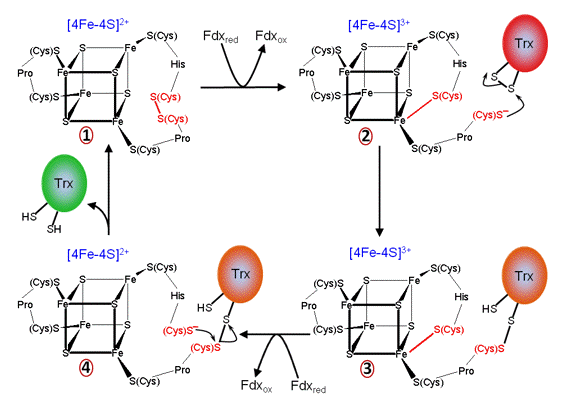
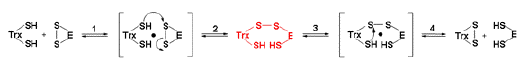
Figure 5. Mechanism of Trx reduction by FTR. The first electron, delivered by Fdx and transmitted through the [4Fe-4S] cluster, cleaves the active-site disulfide, and produces a surface-exposed thiolate and a cysteine-based thiyl radical (1→2). This radical becomes stabilized by covalent attachment of the cysteine to the closest Fe atom of the cluster, thereby forming an oxidized (3+), five-coordinate cluster. The surface exposed thiolate then acts as a nucleophile, and attacks the disulfide bond of a Trx, forming the one-electron-reduced heterodisulfide intermediate (2→3). This reaction anchors a Trx molecule to a docking area designed to fit the protein. The Fdx docking area on the opposite side of FTR becomes free for a second interaction with reduced Fdx, which transfers a second electron to the [4Fe-4S] cluster. This transfer results in the reduction of the cluster-ligated Cys, and reestablishes the original 2+ oxidation state of the cluster (3→4). The newly reduced internal Cys attacks and cleaves the heterodisulfide linkage between FTR and Trx, thus releasing reduced Trx. In a last step (4→1), the closing of the active-site disulfide completes the reaction cycle.
The reduced Trx, in turn, reduces the regulatory disulfide of the target protein by a thiol-disulfide exchange reaction. Such reactions with Trx also proceed via the formation of a transient mixed disulfide between Trx and the target protein, shown in red in the scheme below.
Target Proteins of the Ferredoxin/Thioredoxin System
Most well studied light-regulated enzymes are members of the Calvin-Benson cycle. This cyclic pathway of CO2 incorporation can be divided into three phases: carboxylation, reduction, and regeneration (Figure 6).
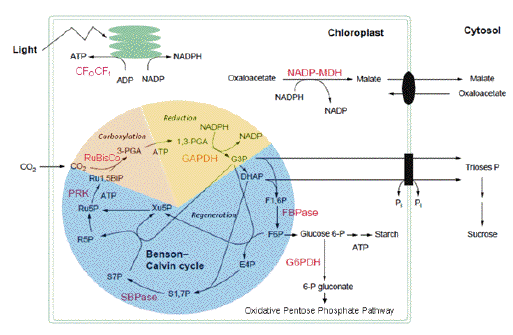
Figure 6. Calvin-Benson cycle and associated light-regulated enzymes. (Modified from Ruelland & Miginiac-Maslow 1999)
In the dark, light control mechanisms switch off the carboxylation and reduction phases, and disconnect the regeneration phase by turning off three enzymes catalyzing irreversible reactions, two positioned early in the regeneration phase (fructose-1,6-bisphosphatase and sedoheptulose-1,7-bisphosphatase), and a third at the end (phosphoribulokinase). Thus, the energy-demanding reactions of the cycle are turned off, while enzymes catalyzing reversible reactions of the regeneration phase that are needed for carbohydrate transformations in dark metabolism remain active. Further, in contrast to members of the Calvin-Benson cycle, the first enzyme of the oxidative pentose phosphate pathway, glucose 6-phosphate dehydrogenase, which provides reducing equivalents for dark metabolism, responds to light in an opposing manner, i.e., oxidative activation in the dark and reductive deactivation in the light.
ATP-synthase and NADP-malate dehydrogenase, two enzymes associated with CO2 assimilation, are also light-controlled. Whereas ATP-synthase provides the ATP needed for the Calvin-Benson cycle, NADP-malate dehydrogenase of C3 plants participates in a shuttle system by exporting surplus reducing equivalents from the chloroplast to the cytoplasm, thereby maintaining a favorable redox equilibrium in the plastid. In certain C4 plants, NADP-malate dehydrogenase is a member of a CO2 concentrating mechanism and is, therefore, directly involved in CO2 assimilation. Enzymes involved in lipid biosynthesis, starch and nitrogen metabolism have also been found to be regulated by the Fdx/Trx system in chloroplasts (Schürmann & Buchanan, 2008). In the past decade, proteomic results have greatly expanded the list of putative targets in chloroplasts and other parts of the plant (Buchanan & Balmer, 2005). While a number of these candidates have been shown to be authentic targets, most still await confirmation.
All enzymes targeted by Trx have at least one regulatory disulfide bridge, which is absent in cytoplasmic counterparts. The regulatory Cys are not embedded in a conserved consensus motif, and are separated by a variable number of residues, sometimes located on a loop structure absent from non-light-regulated isoforms. The mechanisms by which the enzyme activities are light-modulated are as diverse as the regulatory disulfide motifs. These observations suggest that light regulation has evolved individually for each target enzyme.
Target Enzymes in the Calvin-Benson Cycle
Phosphoribulokinase (PRK) is the last enzyme of the regeneration phase and is unique to the Calvin-Benson cycle. In presence of Mg2+ PRK catalyzes the ATP-dependent phosphorylation of ribulose 5-phosphate to ribulose-1,5-bisphosphate, the acceptor molecule for the carboxylation reaction. This homodimeric protein of ~80 kDa contains a redox-sensitive disulfide bridge formed between two Cys separated by ~40 residues, which are proposed to be located on a loop involved in binding ATP (Harrison et al., 1998) close to the N-terminus of each subunit. Neither of the two Cys is directly involved in binding ATP, but formation of the disulfide in the oxidized enzyme perturbs the ATP binding site, and inactivates the enzyme. PRK appears to be the only known Trx-dependent enzyme with a regulatory Cys as part of its active site.
Moreover, in the dark PRK is further deactivated by forming aggregates with glyceraldehyde-3-phosphate dehydrogenase and the small chloroplast protein CP12. In the light, the enzyme is released and activated most efficiently by reduced Trx f (see below) (Marri et al., 2009).
Ribulose-1,5-bisphosphate-carboxylase/oxygenase (RuBisCo), the sole enzyme of the carboxylation phase and also unique to the Calvin-Benson cycle, catalyzes the addition of CO2 and H2O to ribulose-1,5-bisphosphate to yield two molecules of 3-phosphoglycerate. In a side reaction, RuBisCo uses O2 instead of CO2 to produce one molecule each of 3-phosphoglycerate and 2-phosphoglycolate. This oxygenase activity drains carbon from the cycle that is partly recovered by photorespiration in peroxisomes and mitochondria. Oxygenation proceeds more slowly than carboxylation, and its magnitude depends on the relative amounts of O2 and CO2 in the environment. RuBisCo has long been known to be activated by light that is due in part to increasing Mg2+ concentration and alkalization of the stroma. In certain plant species light activates additionally via the Fdx/Trx system, however, not directly the enzyme, but via RuBisCo activase, an enzyme that in most plants consists of two isoforms. Following reductive activation by Trx f, the activase dissociates and, in an ATP requiring reaction, removes a wide variety of inhibitory sugar phosphates from the active site of RuBisCo, thereby restoring full enzyme activity. The larger of the two isoforms contains in a C-terminal extension (CTE) two Cys separated by 18 residues that form a redox-active disulfide. Through reduction of this disulfide, the sensitivity of the activase to ADP inhibition is greatly diminished, and its activity is increased. The redox changes in the larger isoform, also alter the activity of the smaller isoform presumably through cooperative interactions (Figure 7).
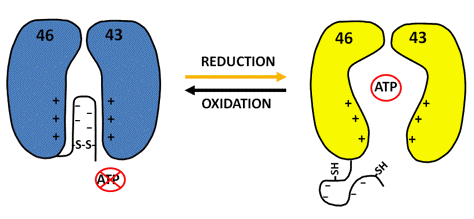
Figure 7. Model for activation of RuBisCo activase. Experimental results suggest that the disulfide carrying the CTE is located near the ATP binding site and selectively interferes with ATP (but not ADP) binding and ATP hydrolysis. Reduction of the disulfide by Trx f is accompanied by a conformational change, probably rendering the reduced extension more flexible. This change removes the CTE blocking the ATP binding site, a mechanism reminiscent of the situation observed for glyceraldehyde-3-phosphate dehydrogenase and NADP-dependent malate dehydrogenase (see below).
Glyceraldehyde-3-phosphate-dehydrogenase (GAPDH) catalyzes the freely reversible reduction of 1,3-bisphosphoglycerate to glyceraldehyde 3-phosphate in the presence of NAD(P)H in the sole reductive step of the Calvin-Benson cycle. This enzyme, which is specific to photosynthetic tissue, exhibits dual cofactor specificity toward pyridine nucleotides with a kinetic preference for NADP(H). This activity is regulated by metabolites, reduction via Trxs, and reversible aggregate formation. The NADH-dependent activity of the enzyme, needed for chloroplast dark metabolism, is constitutive and insensitive to any kind of regulation.
GAPDH is found in two isoforms with different regulatory properties, a minor homotetrameric A4-isoform, known as the non-regulatory GAPDH, and a major heterotetrameric A2B2-isoform, modulated by Trx and metabolites. The two types of subunits of the A2B2-GAPDH are quite similar. The major difference is the presence of a flexible CTE of about 30 residues in the B-subunit containing two invariant Cys, and a large number of negatively charged residues. The CTE is highly homologous to the C-terminus of CP12, which participates in the aggregation of non-regulatory GAPDH with PRK.
Redox-dependent association-dissociation appears to be the main mechanism responsible for the light induced activity changes in GAPDH. In the dark both isoforms are found as large aggregates with strongly inhibited NADPH-dependent activity. Upon reduction by Trx f these aggregates dissociate and GAPDH regains full NADPH-linked activity (Figures 8 and 9).
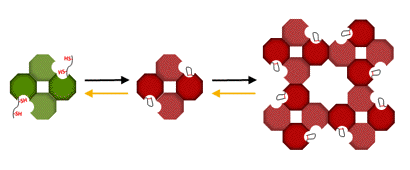
Figure 8. Schematic representation of association-dissociation of A2B2-GAPDH. In the A2B2-isoform the redox-sensitive disulfide is formed by the Cys in the CTE. In the oxidized enzyme, the CTE is positioned in a cleft between subunits, restricting access to the coenzyme binding site and allowing formation of higher oligomers that maintain the enzyme in a kinetically inhibited conformation. Reduction by Trx f dissociates the aggregates, and releases the CTE giving access to the coenzyme binding site and reestablishing full NADPH-dependent activity (Sparla et al., 2002).
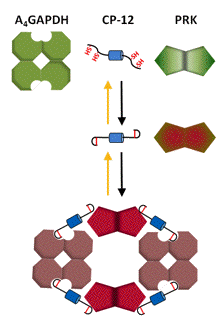
Figure 9. Schematic representation of association-dissociation of A4-GAPDH, CP-12 and PRK. In the homotetrameric A4-isoform, which has no CTE, the small protein, CP12, with its four Cys mimics the CTE. When oxidized, the Cys form two disulfides, one in the N-terminal, and one in the C-terminal region of CP12, which then acts as a linker forming noncovalent complexes between GAPDH and PRK. These complexes aggregate to higher molecular structures keeping both enzymes inactive in the dark. In the light, Trx f reduces the disulfide bonds on CP12 and PRK, releasing both enzymes in active form (Marri et al., 2008, 2009).
Fructose-1,6-bisphosphatase (FBPase) hydrolyzes fructose-1,6-bisphosphate to fructose-6-phosphate and inorganic phosphate. The reaction has a high negative free energy, making it irreversible, and an important point of regulation in the beginning of the regeneration phase. FBPase is a homotetramer of 160 kDa, very similar in primary structure to its cytoplasmic counterpart, however, containing an insert in the middle of the primary sequence of each subunit. This insert forms a flexible loop and contains three cysteines, two of them separated by ~18 residues, which form the regulatory disulfide (Figure 10).
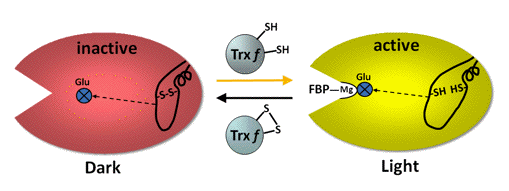
Figure 10. Schematic representation of the activation mechanisms of FBPase. In the oxidized enzyme the disulfide bond locks the 20 Å distant active site, through the movement of two ß-strands, in an inactive configuration that does not permit binding of essential Mg2+ ions. Reduction by Trx f only loosens the loop structure and moves through the ß-strands a glutamate back into the active site making it catalytically competent to bind Mg2+ and substrate (Chiadmi et al., 1999; Dai et al., 2000).
In essence, light reduction of the regulatory disulfide has an allosteric effect on the distant active site by greatly decreasing the Mg2+ requirement for efficient catalysis (Balmer et al., 2001). This, in concert with the alkalization and the increase of available Mg2+ in the stroma, greatly boosts FBPase activity.
Sedoheptulose-1,7-bisphosphatase (SBPase) is the third of the three enzymes unique to the Calvin-Benson cycle. SBPase catalyzes the hydrolysis of sedoheptulose-1,7-bisphosphate, another irreversible step early in the regeneration phase of the cycle. Activity of the enzyme strictly depends on reduction by Trx f and is further regulated by stromal pH and Mg2+ level as well as by substrate and the products of the reaction (Schürmann & Buchanan 2008). A homodimer of 70 kDa, SBPase contains a regulatory disulfide bridge in the N-terminal region with the two Cys separated by four residues. The active site is located in the C-terminal part of the primary sequence. Based on homology modeling the disulfide is situated in a loop, which might function similarly as observed for FBPase, i.e., by acting on nearby ß-strands that modify the conformation of the distant catalytic site (Chiadmi et al., 1999).
Additional Chloroplast Target Enzymes
ATP-synthase, the chloroplast coupling factor CFOCF1, is not a member of the Calvin-Benson cycle, but provides ATP needed for the kinase reactions of the cycle. This enzyme is a membrane protein complex composed of the integral membrane portion CFO and the hydrophilic CF1 consisting of five subunits. It uses the proton-motive force across the thylakoid membrane to synthesize ATP from ADP and Pi. ATP synthase is a latent enzyme, and its activity is regulated in vivo by the transmembrane electrochemical proton gradient inducing conformational rearrangements (Ort & Oxborough, 1992). The potential gradient thus acts as a driving force for phosphorylation, and as an activator for the reversible conversion of the complex to catalytic competence.
In addition to this electrochemical activation, ATP synthase is subject to redox regulation by the Fdx/Trx system. The structural element allowing for thiol modulation is comprised of two Cys separated by five residues in the CF1
 -subunit, forming a disulfide bond in the oxidized enzyme. Reduction of CF1 is very rapid, even in weak light, probably due to the positive redox potential of the disulfide, thus permitting a higher rate of ATP formation at limiting electrochemical potential. Trx-linked regulation also allows the enzyme to be switched off in the dark to avoid wasteful ATP hydrolysis (Samra et al., 2006; Wu et al., 2007). This is in line with the observation that in organisms such as cyanobacteria, CF1 lacks the regulatory disulfide segment seen in the chloroplast counterpart. In these organisms, CF1 is required for both photo- and oxidative-phosphorylation and, therefore, should not be turned off in the dark.
-subunit, forming a disulfide bond in the oxidized enzyme. Reduction of CF1 is very rapid, even in weak light, probably due to the positive redox potential of the disulfide, thus permitting a higher rate of ATP formation at limiting electrochemical potential. Trx-linked regulation also allows the enzyme to be switched off in the dark to avoid wasteful ATP hydrolysis (Samra et al., 2006; Wu et al., 2007). This is in line with the observation that in organisms such as cyanobacteria, CF1 lacks the regulatory disulfide segment seen in the chloroplast counterpart. In these organisms, CF1 is required for both photo- and oxidative-phosphorylation and, therefore, should not be turned off in the dark.
Glucose-6-phosphate-dehydrogenase catalyzes the first committed step of the oxidative pentose phosphate pathway, the oxidation of glucose 6-phosphate to 6-phosphogluconate with concomitant reduction of NADP. The chloroplast isoform is strictly light regulated by the Fdx/Trx system, but in contrast to enzymes of the Calvin-Benson cycle, is deactivated by reduction in the light, and activated by oxidation in the dark. This response is essential in providing for differential control of the two opposing carbon pathways in the chloroplast, minimizing the simultaneous occurrence of carbohydrate synthesis (via the Calvin-Benson cycle) and degradation (via the oxidative pentose phosphate pathway). In land plants the two Cys engaged in the regulatory disulfide bridge are found in a nine residue motif, located in the N-terminal half of the protein near the NADP-binding domain and active site. Based on homology modeling, the regulatory disulfide motif is part of an exposed loop, accessible to Trx. Whereas Trx m has been thought to be the unique regulatory protein, recent results show that reduced Trx f is equally efficient in deactivating the enzyme (Nee et al., 2009).
NADP-dependent malate dehydrogenase (NADP-MDH) is a chloroplast enzyme present in green algae and different types of land plants. The enzyme catalyzes the reduction of oxaloacetate to malate using NADPH as reductant in a reaction that strictly requires light-activation via the Fdx/Trx system. In C3 plants, NADP-MDH functions in a shuttle mechanism exporting surplus reducing equivalents in the form of malate from chloroplasts to the cytosol, thereby helping to maintain a favorable redox equilibrium in the stroma. In C4 plants of the NADP-malic enzyme type (maize, sugarcane or sorghum), NADP-MDH functions as a member of a carbon trapping and transport system of mesophyll cells. It reduces oxaloacetate, the first CO2 fixation product that has been imported in the chloroplast after formation from phosphoenolpyruvate and CO2 in the cytosol. The newly formed malate is then transported into chloroplasts of the bundle-sheath cells where it is decarboxylated by malic enzyme, thereby releasing CO2 and generating NADPH. The CO2 is fixed by RuBisCo, and the NADPH is used in the Calvin-Benson cycle, together with ATP generated by photophosphorylation.
A homodimer of 85 kDa, chloroplast NADP-MDH differs from its NAD-dependent cytosolic homolog by the presence of Cys-containing N- and C-terminal extensions. In the oxidized enzyme, these Cys form two disulfide bridges, one within the N-terminal extension, and one between a Cys on the C-terminal extension and a Cys in the core of the protein. Reduction of either disulfide has a differential effect on enzyme activity, but only a reduction of both disulfides provides maximal activity. This can be understood on the basis of the molecular structure (Figure 11).

Figure 11. Schematic representation of the activation of MDH. The highly flexible N-terminal extension is located at the interface between subunits, where it makes a number of mainly hydrophobic contacts with both the catalytic domain of one subunit, and the coenzyme-binding domain of the other. The reduction of the disulfide is thought to relax this rigid structure, and free the catalytic domain to adopt a productive conformation. The C-terminal extension of the oxidized enzyme is held against the core structure by the disulfide bond, and shields the entrance to the active site by occupying the position of the natural substrate oxaloacetate. Negative charges at the tip of the extension, a penultimate glutamate and the C-terminal carboxylate, are instrumental in this inhibitory action by mimicking the dicarboxylic substrate. The negative charges can also interact with bound NADP+ and, in this way, inhibit the enzyme. Trx reduction of the C-terminal disulfide destabilizes the extension rendering it very mobile thereby permitting access to the active site (Miginiac-Maslow & Lancelin, 2002).
Trxs f and m are both capable of activating NADP-MDH, but as Trx f is more efficient under certain in vitro conditions, it may be the primary activator (Schürmann & Jacquot 2000; Schürmann & Buchanan, 2001).
Redox Potentials and Target Enzyme Activation
The redox potential difference between Fdx and Trxs indicates that Trx reduction is thermodynamically very favorable (Table 1).
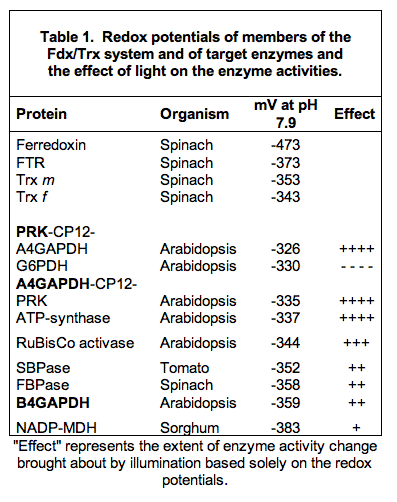
A comparison of the redox potentials of the target enzymes further suggests an order in the action of Trxs on the targets upon illumination. The group of targets with the most positive redox potentials will be reduced very rapidly, thereby turning off the oxidative pentose phosphate pathway, and preparing acceptor molecules for carboxylation. This rapid-response group includes G6PDH, the aggregates of PRK and non-regulatory GAPDH linked by CP12 and ATP-synthase. Then the activation of RuBisCo via RuBisCo activase starts carboxylation, and, finally, the activation of the two phosphatases and GAPDH (regulatory form) enables regeneration of the CO2 acceptor. Depending on the electron pressure, different redox equilibria can be reached, resulting in defined degrees of activation. These equilibria could be further modified by effector molecules that allow fine-tuning of the enzymes.
The redox potential of NADP-MDH is more negative than that of the Trxs, thus requiring an excess of reduced Trx for activation. This requirement is consistent with the role of the enzyme in C3 plants, where it transports excess reducing equivalents from the chloroplast to the cytosol (see Figure 6).
Concluding Remarks
Experiments with algal cells and chloroplasts initiated in the 1960s provided evidence that light not only supplies energy for CO2 assimilation, but also is perceived as a regulatory signal that modifies the activity of chloroplast enzymes (Buchanan et al., 2002; Bassham, 1971). This function enables chloroplasts to switch from dark to light metabolism and to adjust enzyme activities in response to changing light conditions. Research in subsequent decades has established the elements involved in transmitting the light signal to enzymes, leading to the discovery of Trxs in plants and to their involvement in enzyme regulation, work that has given birth to the field of redox biology. The redox-based mechanism by which this occurs in oxygenic photosynthesis has become known as the Fdx/Trx system of enzyme regulation. Research in recent years has added structural and mechanistic details, which have provided a better understanding of the mechanism of signal transfer via this mechanism. Future studies should explore the Trx-linked enzymes that have been identified in proteomic studies, and further elucidate details of the changes that proteins undergo when regulated by redox.
References
Arnér ESJ, Holmgren A (2000) Physiological functions of thioredoxin and thioredoxin reductase. Eur J Biochem 267: 6102-6109
Balmer Y, Stritt-Etter A-L, Hirasawa M, Jacquot J-P, Keryer E, Knaff DB, Schürmann P (2001) Oxidation-reduction and activation properties of chloroplast fructose-1,6-bisphosphatase with mutated regulatory site. Biochemistry 40: 15444-15450
Bassham JA (1971) Photosynthetic carbon metabolism. Proc Natl Acad Sci USA 68: 2877-2882
Buchanan BB (1980) Role of light in the regulation of chloroplast enzymes. Annu Rev Plant Physiol 31: 341-374
Buchanan BB, Schürmann P, Wolosiuk RA, Jacquot J-P (2002) The ferredoxin/thioredoxin system: from discovery to molecular structures and beyond. Photosynth Res 73: 215-222
Buchanan BB, Balmer Y (2005) REDOX REGULATION: A Broadening Horizon. Annu Rev Plant Biol 56: 187-220
Chiadmi M, Navaza A, Miginiac-Maslow M, Jacquot J-P, Cherfils J (1999) Redox signalling in the chloroplast: structure of oxidized pea fructose-1,6-bisphosphate phosphatase. EMBO J 18: 6809-6815
Dai S, Friemann R, Glauser DA, Bourquin F, Manieri W, Schürmann P, Eklund H (2007) Structural Snapshots along the Reaction Pathway of Ferredoxin-Thioredoxin Reductase. Nature 448: 92-96
Dai S, Schwendtmayer C, Johansson K, Ramaswamy S, Schürmann P, Eklund H (2000) How does light regulate chloroplast enzymes? Structure-function studies of the ferredoxin/thioredoxin system. Quart Rev Biophys 33: 67-108
Glauser DA, Bourquin F, Manieri W, Schürmann P (2004) Characterization of ferredoxin:thioredoxin reductase (FTR) modified by site-directed mutagenesis. J Biol Chem 279: 16662-16669
Gontero B, Avilan L, Lebreton S (2006) Control of carbon fixation in chloroplasts. Annual Plant Reviews 22: 187-218
Harrison DH, Runquist JA, Holub A, Miziorko HM (1998) The crystal structure of phosphoribulokinase from Rhodobacter sphaeroides reveals a fold similar to that of adenylate kinase. Biochemistry 37: 5074-5085
Hase T, Schürmann P, Knaff DB (2006) Ferredoxin and ferredoxin-dependent enzymes. Advances in Photosynthesis and Respiration 24: 477-498
Marri L, Trost P, Trivelli X, Gonnelli L, Pupillo P, Sparla F (2008) Spontaneous assembly of photosynthetic supramolecular complexes as mediated by the intrinsically unstructured protein CP12. J Biol Chem 283: 1831-1838
Marri L, Zaffagnini M, Collin V, Issakidis-Bourguet E, Lemaire SD, Pupillo P, Sparla F, Miginiac-Maslow M, Trost P (2009) Prompt and Easy Activation by Specific Thioredoxins of Calvin Cycle Enzymes of Arabidopsis thaliana Associated in the GAPDH/CP12/PRK Supramolecular Complex. Mol Plant 2: 259-269
Meyer Y, Buchanan BB, Vignols F, Reichheld JP (2009) Thioredoxins and Glutaredoxins: Unifying Elements in Redox Biology. Annu Rev Genet 43: 335-367
Meyer Y, Siala W, Bashandy T, Riondet C, Vignols F, Reichheld JP (2008) Glutaredoxins and thioredoxins in plants. Biochim Biophys Acta 1783: 589-600
Miginiac-Maslow M, Lancelin J-M (2002) Intrasteric inhibition in redox signalling: Light activation of NADP-malate dehydrogenase. Photosynth Res 72: 1-12
Mora-Garcia S, Stolowicz FG, Wolosiuk RA (2006) Redox signal transduction in plant metabolism. Annual Plant Reviews 22: 150-186
Nee G, Zaffagnini M, Trost P, Issakidis-Bourguet E (2009) Redox regulation of chloroplastic glucose-6-phosphate dehydrogenase: a new role for f-type thioredoxin. FEBS Letters 583: 2827-2832
Ort DR, Oxborough K (1992) In situ regulation of chloroplast coupling factor activity. Annu Rev Plant Physiol Plant Mol Biol 43: 269-291
Ruelland E, Miginiac-Maslow M (1999) Regulation of chloroplast enzyme activities by thioredoxins: activation or relief from inhibition? Trends Plant Sci 4: 136-141
Samra HS, Gao F, He F, Hoang E, Chen Z, Gegenheimer PA, Berrie CL, Richter ML (2006) Structural analysis of the regulatory dithiol-containing domain of the chloroplast ATP synthase gamma subunit. J Biol Chem 281: 31041-31049
Schürmann P, Buchanan BB (2001) The structure and function of the ferredoxin/thioredoxin system in photosynthesis. Advances in Photosynthesis and Respiration 11: 331-361
Schürmann P, Buchanan BB (2008) The Ferredoxin/Thioredoxin System of Oxygenic Photosynthesis. Antioxid Redox Signal 10: 1235-1274 Schürmann P, Jacquot J-P (2000) Plant thioredoxin systems revisited. Annu Rev Plant Physiol Plant Mol Biol 51: 371-400
Sparla F, Pupillo P, Trost P (2002) The C-terminal extension of glyceraldehyde-3-phosphate dehydrogenase subunit B acts as an autoinhibitory domain regulated by thioredoxins and nicotinamide adenine dinucleotide. J Biol Chem 277: 44946-44952
Walters EM, Garcia-Serres R, Jameson GNL, Glauser DA, Bourquin F, Manieri W, Schürmann P, Johnson MK, Huynh BH (2005) Spectroscopic characterization of site-specific [Fe4-S4] cluster chemistry in ferredoxin:thioredoxin reductase : Implications for the catalytic mechanism. J Am Chem Soc 127: 9612-9624
Walters EM, Garcia-Serres R, Naik SG, Bourquin F, Glauser DA, Schürmann P, Huynh BH, Johnson MK (2009) Role of Histidine-86 in the Catalytic Mechanism of Ferredoxin:Thioredoxin Reductase. Biochemistry 48: 1016-1024
Wu G, Ortiz-Flores G, Ortiz-Lopez A, Ort DR (2007) A Point Mutation in atpC1 Raises the Redox Potential of the Arabidopsis Chloroplast ATP Synthase
-Subunit Regulatory Disulfide above the Range of Thioredoxin Modulation. J Biol Chem 282: 36782-36789
Xu X, Schürmann P, Chung JS, Hass MAS, Kim SK, Hirasawa M, Tripathy JN, Knaff DB, Ubbink M (2009) Ternary Protein Complex of Ferredoxin, Ferredoxin:Thioredoxin Reductase, and Thioredoxin Studied by Paramagnetic NMR Spectroscopy. J Am Chem Soc 131: 17576-17582
02/14/10