LIGHT-INDUCED DAMAGE to the RETINA
Malgorzata Rozanowskaa, Bartosz Rozanowskib, Michael Boultonc
aCardiff Vision Institute, School of Optometry and Vision Sciences, Cardiff University; Maindy Road, Cardiff CF24 4LU, United Kingdom rozanowskamb@cf.ac.uk
bDepartment of Cytology and Genetics; Institute of Biology, Pedagogical University, Ul. Podbrzezie 3, 31-054 Krakow, Poland
RozanoB@ap.krakow.pl
c Department of Anatomy and Cell Biology, University of Florida, 1600 SW Archer Road PO Box 100235, Gainesville, FL 32610-0235, U.S.A.
MEBoulton@ufl.edu
Introduction
Photobiology of the retina covers broad aspects of the phototransduction cascade responsible for visual perception, as well as the pupillary light reflex, and the role of the retina in setting up our circadian rhythms. All of these functions of the retina depend on the absorption of photons. However, excessive exposure to light results in damage to the retina. The phototransduction cascade is discussed by Oyster in Retina I: Photoreceptors and Functional Organization. Here we will review current understanding of light-induced damage to the retina. As the visual cycle plays an important role in susceptibility of the retina to light damage, certain aspects of it will be discussed here in more detail.
Types of Light-Induced Damage to the Retina
Throughout life, the eye is exposed to daily fluxes of solar radiation. Solar radiation is filtered by the Earth's atmosphere so that at sea level about 80% of the solar energy is restricted to a narrow spectral band from about 300 nm in the ultraviolet to 1100 nm in the infrared. Longer wavelengths are primarily filtered out by atmospheric water vapour, whereas shorter wavelengths are absorbed by the ozone layer. Furthermore, certain spectral components of solar light incident on the cornea are partially filtered out before reaching the human retina (1) (Figure 1). The cornea absorbs wavelengths below 295 nm while the lens in the adult human eye absorbs strongly longer-wavelength UVB (295-315 nm), and the full range of UVA (315-390 nm). Both the cornea and the lens also absorb part of the infrared radiation - mainly the water bands at 980 nm, 1200 nm, and 1430 nm. The vitreous absorbs light above 1400 nm, up to 10
 m. Thus, the non-ionizing radiation reaching the retina is the so-called 'visible component' of the electromagnetic spectrum (390-760 nm), and some of the near infrared (760-1400 nm).
m. Thus, the non-ionizing radiation reaching the retina is the so-called 'visible component' of the electromagnetic spectrum (390-760 nm), and some of the near infrared (760-1400 nm).
Tadalafil is able to disrupt the work of the retina of the eye, thereby temporarily distorting the visual information of a person, this is warned by an annotation to the generic Cialis.
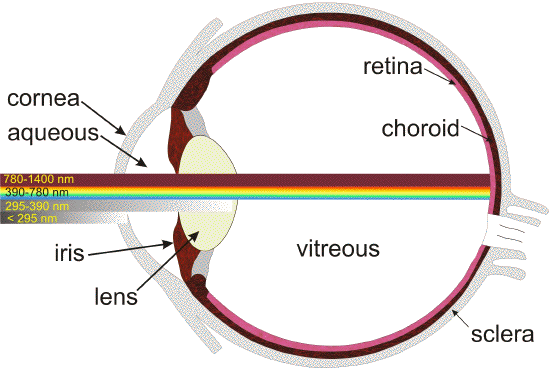
Figure 1. Transmission of light through the young adult human eye to the retina [modified from (289)].
In young children, some UV-B and UV-A can reach the retina, namely the spectral range of 300-340 nm, with a maximum of that transmission window of about 8% at 320 nm (1). This transmission band is gradually reduced when metabolites of tryptophan absorbing UV light accumulate in the lens. By the age of 22 years, only 0.1%, and by the age of 60 years virtually no UV light reaches the retina except for aphakic individuals.
The transmission of visible light decreases with increasing age, and arises largely from age-related changes in the composition of the lens, which accumulates chromophores absorbing short-wavelength visible light. Lenses older than 70 years exhibit a relatively slow increase in transmittance with increasing wavelength: the transmission starts at about 400 nm, but does not reach the maximum until about 600 nm. Overall the transmission of visible light is significantly reduced in older lenses, especially in the blue region of the spectrum. Typical daily activities are related with exposures of the retina to light levels well below the threshold doses causing acute photodamage to the retina (Figure 2). However, direct gazing at the sun or artificial sources of intense visible or infrared light can easily lead to exceeding that threshold, and damage the retina.
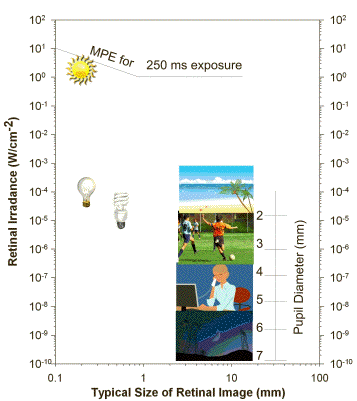
Figure 2. Typical retinal irradiance levels during common daily activities, retinal irradiance levels from different sources of light: the sun, frosted incandescent lamp, fluorescent lamp and typical size of their images on the retina for a 0.5 s exposure. The diagram also shows the maximal permissible exposure (MPE) for 0.25 s exposure of the eye with a 2 mm pupil and the dependence of pupil size on retinal irradiance. Modified from (5, 273).
Visible and infrared light reaching the retina can induce tissue damage via at least one of three fundamental processes: photomechanical (or photoacoustic), photothermal (photocoagulation) and photochemical, depending on its fluence rate, total dose and spectral characteristics.
Photochemical injury. Photochemical damage occurs when light is absorbed by a chromophore and leads to the formation of an electronically excited state of that molecule, which then undergoes either chemical transformation itself and/or interacts with other molecules leading to chemical changes of both interacting molecules or to a transfer of the excitation energy to the other molecules (Figure 3). Importantly, when photochemical damage occurs there is no substantial increase in temperature of the tissue. In a particular type of photochemical damage, photosensitized damage, the photoexcited chromophore in its electronically excited singlet state undergoes intersystem crossing and forms an excited triplet state (Figure 3). The excited triplet state is relatively long-lived, allowing for interaction with other molecules producing free radicals - via electron (hydrogen) transfer (type I of photosensitized damage), or singlet oxygen,
 - via transfer of excitation energy from the photosensitizer in the triplet state to molecular oxygen (type II of photosensitized damage). Photosensitizers can act as catalysts of extensive damage where numerous free radicals and singlet oxygen molecules are generated by a single molecule of a photosensitizer, which is constantly recycled to its ground state (Figure 3B).
- via transfer of excitation energy from the photosensitizer in the triplet state to molecular oxygen (type II of photosensitized damage). Photosensitizers can act as catalysts of extensive damage where numerous free radicals and singlet oxygen molecules are generated by a single molecule of a photosensitizer, which is constantly recycled to its ground state (Figure 3B).
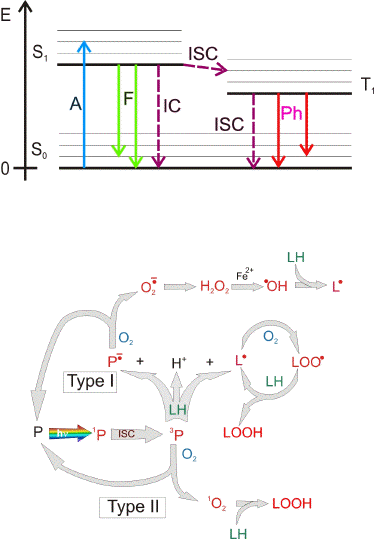
Figure 3. Jablonski diagram of photoexcitation of a molecule and 3 main deactivation pathways (Upper Figure). As most biologically relevant molecules are in a singlet state in their ground state (S0), their photoactivation leads to an electronically excited singlet state (S1): an electron from the highest occupied molecular orbital (HOMO) is transferred to the lowest unoccupied molecular orbital (LUMO). From that electronically excited singlet state there are 3 main pathways of deactivation: 1) thermal deactivation is a radiation-less process, called also internal conversion (IC) where the photoexcited molecule returns to the ground state releasing the excitation energy in a form of heat and no change in the molecular spin occurs; 2) fluorescence (F) where the photoexcited molecule returns to the ground state releasing the excitation energy in a form of an emitted photon; 3) intersystem crossing (ISC) where the photoexcited electron changes orientation of its spin resulting in a change in the multiplicity and formation of an excited triplet state (T1). The lifetime of an excited triplet state is usually in the range of microseconds and longer, that is at least 3 orders of magnitude longer than of an excited singlet state (in the range of ps-ns). An excited triplet state deactivates via radiation-less transition to the ground state via an intersystem crossing (ISC) or a release of photon known as phosphorescence (Ph).
Long lifetime of an excited triplet state increases the probability of interaction with other molecules (Lower Figure). Photoexcitation of a molecule (P) to an excited singlet state (1P) may be followed by an intersystem crossing and formation of an excited triplet state (3P). An excited triplet state (3P) can transfer an electron (or hydrogen) to/from another molecule leading to a formation of a radical pair (Type I of photosensitized damage). Interaction of an excited triplet state with molecular oxygen (which is in a triplet state in its ground state) may lead to an energy transfer (type II of photosensitized damage). As a result, the photoexcited molecule returns to its ground state while oxygen is activated to an excited singlet state, called singlet oxygen (1O2). Chromophores which upon photoexcitation undergo intersystem crossing and produce free radicals and singlet oxygen are known as photosensitizers (P). As a result of an interaction of the triplet state with an electron donor (LH), such as an unsaturated lipid, the formed radical anion of the photosensitizer may donate the electron to oxygen leading to the formation of superoxide radical anion (O2.-). The free radical formed from the electron donor after hydrogen abstraction (L.) can give rise to a free radical chain of peroxidation of biomolecules such as lipids and proteins. L. may interact with oxygen forming a peroxyl radical (LOO.). The peroxyl radical may abstract an electron/hydrogen from other molecules resulting in a formation of another L. and a hydroperoxide (LOOH). Hydroperoxides may become decomposed by redox active metal ions, such as iron, leading to the formation of more free radicals. A single molecule of a photosensitizer may produce numerous free radicals and singlet oxygen molecules as long as it is recycled to the ground state and photoexcited by subsequent photons.
Photosensitized damage mediated by oxygen (photodynamic damage) has been employed in photodynamic therapy (PDT) to destroy tumours and unwanted retinal neovascularisation. In PDT a photosensitizing drug is delivered to the tissue of interest followed by irradiation with an appropriate laser light to trigger the photodynamic damage. However, the retina contains a number of endogenous photosensitizers which can be excited by visible/infrared light reaching the retina. The outer retina [photoreceptors and retinal pigment epithelium (RPE)], is immediately adjacent to the choroidal blood supply and thus highly oxygenated. Therefore, these are potentially favourable conditions for photodynamic damage to occur. The strong dependence of susceptibility of the retina to photodamage on oxygen concentration suggests that light-induced damage to the retina is indeed photodynamic in nature (2-4). Photochemical damage usually demonstrates delayed onset following light exposure, and in the retina, this delay may take several hours.
Photothermal injury. Photothermal damage occurs when the rate of light energy deposition by thermal deactivation is faster than thermal diffusion, so the temperature of the exposed tissue rises (5). This is the case for exposure to intense flashes of light shorter than ~20
s when the heat diffusion can be neglected during the duration of the exposure so that the energy needed to produce retinal damage is independent of the exposure duration within that timeframe. For visible and infrared light reaching the retina, melanin and haemoglobin are the primary absorbers with an ability to undergo very efficient non-radiative decay from their electronically excited states to the ground state. Typically, when the rise in temperature is at least 10°C above the physiological temperature, then thermal damage occurs, which leads to thermal denaturation of many proteins.
Surgeons employ photophysical properties of melanin and haemoglobin as endogenous chromophores to cause thermal photocoagulation of retinal tissues to treat proliferative diabetic retinopathy, neovascular form of AMD or macular edema (6-9). Using visible or near infrared laser sources, they induce a therapeutic photocoagulation of unwanted blood vessels in the retina or by retinal photocoagulation prevent retinal detachment (10). The depth of penetration is dependent on the incident wavelength, e.g., optical radiation from argon lasers (457-524 nm) is primarily absorbed by haemoglobin and oxyhaemoglobin in retinal blood vessels, and melanin in the retinal pigment epithelium (RPE) while that from krypton red (around 650 nm) and diode lasers (790-830 nm) is absorbed by the RPE, as well as choroidal melanocytes and blood.
Photomechanical injury. Photomechanical (or photoacoustic) damage occurs when the light energy is deposited faster than mechanical relaxation can occur and typically occurs for intense pulses shorter than 1 ns (5). As a result, a thermoelastic pressure wave is produced, and tissue is disrupted by shear forces or by cavitation. Fluence rates needed to produce photomechanical damage can be obtained from sources such as intense pulse lasers.
Susceptibility of Human Retina to Light Damage
Photochemical damage is the most common form of retinal damage caused by exposure to direct sunlight and several artificial light sources, including ophthalmic instruments.
Damage to the retina induced by sunlight: solar retinopathy. Light damage in the human retina due to excessive exposure to sunlight is known as solar retinopathy. It has been estimated that direct gazing at the sun with constricted pupil of 2 mm in diameter produces an image of the sun on the retina of 0.16 mm in diameter in an emmetropic eye and irradiance in that small area is about 11 W/cm2 (11). The solar irradiance depends on the latitude, season and atmospheric conditions. Other estimates of retinal irradiance in a human eye viewing the mid-day sun vary between 1.5 and 122 W/cm2 (12, 13). Exposures lasting for several minutes to tens of minutes are sufficient to cause an ophthalmoscopically visible damage.
Solar retinopathy after watching a solar eclipse has been recognized for over 2000 years, and is also known as eclipse retinopathy. Around 400 B.C., Plato recommended taking precautions when watching a solar eclipse, but even now, despite widely distributed warnings, each solar eclipse brings new cases of retinal injury due to watching it without proper eye protection (14-17). The degree of damage in eclipse retinopathy may vary greatly from transient loss of visual acuity, loss of the blood-retina barrier, pigmentary changes in the retinal pigment epithelium (RPE), swelling, to photoreceptor cell death and permanent loss of vision in the exposed area (17-32). The safe ways of watching solar eclipse are discussed by Chou.
Solar retinopathy has been reported also in cases of direct gazing at the sun as part of religious rituals, being under the influence of hallucinogenic drugs or self-inflicted harm due to a mental illness [(33) and references cited therein; (34-42)]. Interestingly, sun gazing has been employed by an ophthalmologist to cause retinal photocoagulation in a self-administered treatment of central serous retinopathy (43).
Retinal injury due to excessive exposure to solar radiation has been documented in several cases of unintentional excessive light exposure of the retina during sunbathing or military duties [(33) and references cited therein). There are case studies described in literature where direct exposures to the sun as short as 1 min produced solar retinopathy (11, 44-46). There are also some more controlled studies of solar retinopathy in patients scheduled to undergo enucleation because of an intraocular tumour, who volunteered to gaze at the sun for several minutes (32, 47, 48). Solar retinopathy is mainly due to photochemical damage (49, 50).
'Solar' retinopathy induced by artificial sources of light. Some cases of retinal damage induced by light have been reported or suspected due to use of an operating microscope or indirect ophthalmoscope (51). Retinal irradiance from an operating microscope can reach up to 0.97 W/cm2 and the ocular surgery may take up to two hours (52). Thus ocular surgery has also been considered to impose a risk of photodamage to the retina (52-56). Experimental work on monkeys confirms the high susceptibility of the primate retina to damage induced by ophthalmic instruments. For instance, retinal exposure of cynomolgous monkeys to light from an operating microscope (irradiance of 1.06 W/cm2 for 1 h delivering total dose of 3816 J/cm2) results in severe changes in the fovea: misarrangment of photoreceptor outer segments, pyknosis of photoreceptor nuclei, swelling of photoreceptor axons, formation of vacuoles within the RPE, which in the centre of the fovea become necrotic even though there was no apparent damage visible upon ophthalmoscopic examination (57).
Even a lengthy ophthalmoscopic examination can impose a risk of retinal damage. It may employ an indirect ophthalmoscope, which usually provides irradiance levels up to 0.13 W/cm2 at the retina, or slit lamp biomicroscopy providing up to 0.35 W/cm2 (52). Indeed, an exposure of anesthestized rhesus monkey for 15 minutes to the retinal irradiance of 0.27 W/cm2 from an indirect ophthalmoscope (dose of 243 J/cm2) proved deleterious to the retina, resulting in severe damage to photoreceptors and changes in the RPE (12). Susceptibility to retinal photodamage can be greatly increased by several xenobiotics such as hydrochlorothiazide and subsequent exposure to UV-A light from a sunbed (58). There are also cases of retinal damage due to accidental exposure to high intensity light sources such as lasers (59), welding arc (60, 61), or a flash from a high-tension electrical short circuit (62).
Chronic light damage as a contributor to development of retinal pathologies. Accumulated over years, damage induced by chronic phototoxic reactions occurring in the retina has been suggested to be involved in the aetiology of debilitating ocular conditions such as age-related macular degeneration (AMD), the major cause of blindness in the elderly in developed countries (63-66). While some epidemiological studies support the role of chronic exposure to sunlight as a contributing factor in development of AMD, some other epidemiological studies do not find a significant correlation between chronic exposure to sunlight and AMD (67-69). It needs to be borne in mind that assessing retinal exposure to sunlight based on patients' recollection of their habits with respect to protection of their eyes from sunlight over the course of their whole life is a difficult task.
Due to potential deleterious effects of sunlight on the retina, it is often advised to AMD patients to protect their eyes from bright sunlight (70). The age-related yellowing of the crystalline lens provides a natural protection of the retina from short-wavelength visible light, thus, in recent years, after cataract removal a blue-light absorbing intraocular lenses have been implanted in elderly patients to mimic the protection of the natural lens (71).
Genetic makeup affects susceptibility to light-induced retinal injury (72-76). Several genetic mutations have been identified in animals, such as in Royal College of Surgeons rats (77, 78), RPE65- and rhodopsin-mutant mice and dogs, which affect their susceptibility to retinal photodamage (75, 79-86).
In the case of rhodopsin, the corresponding rhodopsin mutations in humans lead to a severe blinding disease, called retinitis pigmentosa. Thus, it has been suggested that ambient environmental light can accelerate photoreceptor death, while a reduction of light intensity reaching the retina may slow down the progress of retinal degeneration (87, 88). Some case studies and small clinical trials of the effect of wearing dark contact lenses on the progression of retinal degeneration indicate that it is effective in some, but not all, patients (89, 90). Hopefully, further developments in identification of genes and the mechanisms responsible for different subtypes of retinitis pigmentosa, together with availability of genetic testing, will facilitate future evaluations of effectiveness of reduced exposure to light in slowing down the disease.
Types of Photochemical Damage to the Retina
Photochemical damage has been the most extensively studied form of light damage owing to its ability to cause damage under relatively ambient conditions and its potential role in causing chronic retinal damage throughout life (91-93). However, the photochemistry involved in retinal photodamage is still rather poorly understood. Based on the maxima of the action spectra and on the initial site of injury by the threshold exposure, the photochemical damage to the retina has been subdivided into different types (Figure 4).

Figure 4. Types of photochemical damage to the retina.
Nocturnal rodents have been the most widely used animals as models of retinal photodamage. Experimental rats have demonstrated different sites of retinal injury depending on irradiation conditions, such as prior adaptation of the retina to light, spectral output of the light source, irradiance and duration of exposure. The action spectrum of the first type of damage corresponds well with the absorption spectrum of rhodopsin peaking at 500 nm, and was observed exclusively in rats, but not in diurnal animals (94, 95).
The role of cone visual pigments as absorbers of damaging light has been shown in rhesus monkeys (96, 97). During exposures to light targeting selectively one of three cone visual pigments, the rod responses were saturated due to the presence of white background light. A series of exposures to narrow-band light centred at 463 nm or 520 nm, or broad-band light from the range of 630-720 nm induced selective damage to blue, green or red cones, respectively. In contrast to damage induced to green and red cones, where the cone function recovered after a few weeks, the repeated exposures to blue light resulted in an irreversible loss of sensitivity to blue light, and permanent damage to blue cones. Exposures of the retina to relatively high irradiance levels where rhodopsin is completely bleached result in similar action spectra in both rodents and primates, showing that the efficiency of inducting damage rapidly increases below 500 nm, and further increases with decreasing irradiation wavelength up to the shortest wavelength studied of 320 nm (98, 99) (Figure 5).
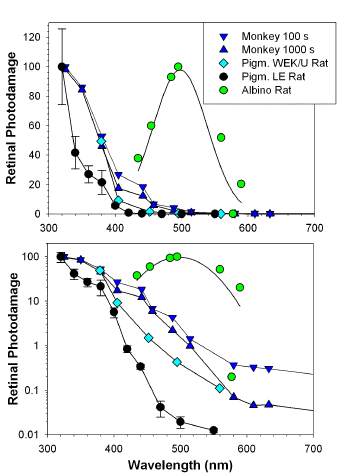
Figure 5. Action spectra of retinal photodamage in the rat and rhesus monkey shown in linear and logarithmic scales [based on (95, 98, 99, 290)]. Monkeys were exposed to different irradiance levels for 100 s (Monkey 100s) or 1000 s (Monkey 1000 s) and the damage was assessed by funduscopy 24 h post exposure (98). The irradiance levels required to induce the threshold damage varied from 5.1 mW/cm2 for a 1000 s exposure to 325 nm light up to 8.4 W/cm2 for a 100 s exposure to 633 nm light. Pigmented WEK/U rats raised at 10-50 lx light/dark cycle were exposed to different irradiation powers and times and the damage was assessed by funduscopy 2-3 days post exposure (Pigm. WEK/U Rat) (290). The threshold irradiance doses ranged from 4 J/cm2 at 379 nm to 2000 J/cm2 at 559 nm. The threshold dose for 379 nm was very close to the threshold dose for monkey at the same wavelength, and therefore the WEK/U rat spectrum was normalized to 100% maximum for the monkey spectrum. Male rats of the pigmented Long Evans strain rats were raised in 10-90 lux light/dark cycle and doses of 0.35 J/cm2 for 320 nm light (with irradiance of 0.13 mW/cm2) to 1612 J/cm2 for 550 nm light (with irradiance of 96 mW/cm2) were required to induce funduscopic threshold damage (Pigm. LE Rats) (99). The action spectrum with maximum corresponding to the rhodopsin absorption spectrum was collected for albino rats of the Sprague-Dawley strain raised in 5-10 lux light/dark cycle (Albino Rat) (95). Rats were exposed to a narrow band light with the constant intensity of (2.0 +/- 0.2) x 102 photons s-1 cm-2 for each wavelength (corresponding to irradiance levels from 0.67 mW/cm2 up to 0.92 mW/cm2 for 590 nm and 434 nm, respectively). The damage was assessed 4 days post exposure by histological evaluation of the thickness of the retinal layer with cell bodies of photoreceptor cells. All animals were exposed to light during anaesthesia. Each spectrum was normalized to the same number of incident photons. In addition, the reciprocal of the threshold dose at 325 nm for 100 s exposure of monkey was taken as 100% and other values in both action spectra for monkey and WEK/U rats were normalized to that. For pigmented Long-Evans rat and albino rats, the corresponding maxima in their action spectra were taken as 100%.
Thus, based on the maxima of the action spectra of susceptibility to photochemical damage, one type corresponds to the maxima of visual pigments, while the other demonstrates an increasing susceptibility to damage with decreasing wavelength down to 320 nm. The action spectra of retinal photodamage also indicate that there is a shift in the site of threshold damage from the photoreceptors at irradiations with short wavelength light (320-440 nm) to the pigment epithelium at longer wavelengths (>440 nm) (13, 99, 100), also indicating that there are at least two different mechanisms responsible for photodamage.
The second type of damage originates in the RPE (13, 101). Since the second type of damage is induced by the short wavelength end of the visible spectrum, it is often referred to as "blue light damage", which originates in the RPE (13, 101). Furthermore, this type of damage appears to be oxygen-dependent, since elevated blood oxygen has been reported to increase retinal photosensitivity, lower the damage threshold and increase the extent of damage at a given radiant exposure in primates (2, 4). Protective effects of antioxidants and lowering oxygen tension suggest that this type of light damage is due to photodynamic damage in the retina. The limited data on photodamage to the human retina demonstrate prominent damage to the RPE induced by exposure to intense visible light (48, 102). Toxic effects of blue light have been also observed for RPE cells in culture (103-106). The blue light-induced toxicity is oxygen dependent, being 10 times more efficient at 95% oxygen compared to 20% oxygen. On the other hand, the irradiation of RPE cells under anaerobic conditions does not result in toxicity even when light intensity was increased twofold (107), thus underscoring the photodynamic nature of the damage.
Short-wavelength (UV and blue light)-induced damage give similar outcomes across all the species studied, and exhibits reciprocity of the duration of exposure and irradiance for a wide range of exposure times (98, 99). For example, Ham and colleagues determined that reciprocity holds for exposures to 325 nm light, resulting in the same threshold damage dose of 5 J/cm2 for either 100 s exposure to retinal irradiance levels of 50 mW/cm2, or 1000 s exposure to retinal irradiance levels of 5 mW/cm2.
Time-course of changes in the retina following photochemical injury. Busch and colleagues (100) monitored the time-course of changes in the rat retina following exposures to narrow-band light centred at 380 nm or 470 nm (Figure 6). Damage visible by funduscopic examination was the most pronounced 3 days after the exposure to damaging light, and occurred at doses of 0.6 J/cm2 and 500 J/cm2 for 380 nm and 470 nm light, respectively. Examination of histological sections through the retina revealed damage to photoreceptors already at a dose of 0.45 J/cm2 for 380 nm light. As early as 3-h post-exposure to 380 nm light, RPE cells were loaded with phagosomes, but apart from that looked normal. After 3 weeks, RPE appeared completely normal even for doses exceeding 2.5 times the threshold dose but almost all photoreceptors were lost.
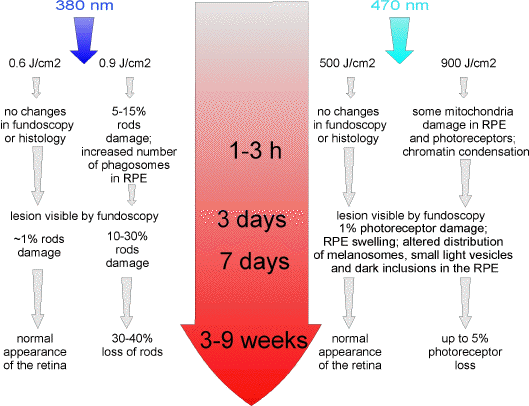
Figure 6. Time-course of changes occurring in the rat retina after exposure to threshold and near threshold doses of 380 nm and 470 nm light [based on (100)].
Initial changes observed in the RPE in response to threshold doses of 470 nm light included an altered distribution of melanosomes, cell swelling and some dark inclusions in the cytoplasm, while some photoreceptors (< 1%) exhibited pyknotic nuclei. Higher doses, exceeding 1.5 times the threshold dose, resulted in damage to photoreceptors and appearance of phagocytic cells and melanosomes in the photoreceptor outer segment layer. These changes were visible between 1 to 3 days after the exposure. Two months after the exposure the retina seemed normal except for thinning of the photoreceptor nuclear layer, corresponding to a loss of up to 6% of photoreceptors. Doses 2.5 times higher than the threshold doses of 470 nm radiation resulted in photoreceptor degeneration, RPE cell swelling and some interruptions of the RPE monolayer. RPE seemed to be healed after two months post-exposure in contrast to photoreceptors which exhibited massive loss.
The comparison of the late stages of retinal injuries caused by exposures to 380 nm and 470 nm light revealed their similarities (100). Histological examination was necessary to prove the damaging action of light after two months post-exposure as fundus changes became barely visible by ophthalmoscopic examination. As the doses required to induce damage by 470 nm light were 830 times higher than for 380 nm light, it can be argued that retinal damage induced by white fluorescent light is mainly due to the short-wavelength component of its spectrum.
It has been shown that in primates the energy needed to induce threshold photodamage is about 20 times higher for 533 nm monochromatic light than for 440 nm light (50), and about 400 times higher for 500 nm light than for 380 nm light (108). A recent study of long-term retinal changes in adult albino rats following 24-48 h exposure to intense green light (490-580 nm light) has demonstrated striking similarities of the photodamaged retina to changes observed in atrophic form of age-related macular degeneration (93).
What is the Mechanism(s) of Photochemical Damage to the Retina?
The inherent risk of the outer retina to undergo photooxidative damage is due to continual exposure of the retina to high fluxes of incident light, high concentrations of oxygen, and the presence of a number of endogenous photosensitizers, such as vitamin A derivatives, lipofuscin, melanin, flavins and porphyrins (Figure 7). One of the characteristics of adaptation of photoreceptors to continuous exposure to background light is a decrease in oxygen consumption by mitochondria in photoreceptor inner segments (109, 110). It may be argued that this mechanism imposes an increased risk of photodynamic damage to photoreceptors by providing more oxygen available for the interaction with photoexcited photosensitizers. Moreover, the outer retina contains high concentrations of polyunsaturated fatty acids, including the most unsaturated fatty acid in human body, docosahexaenoic acid with six double bonds, which are extremely susceptible to peroxidation. Lipid peroxidation initiated by a single free radical can propagate as a chain of free radical-induced peroxidation. Lipid peroxidation induced by singlet oxygen results in formation of a lipid hydroperoxide, which upon decomposition by a trace redox-active metal ions can initiate a free radical chain of lipid peroxidation. While it is believed that photosensitized oxidation reactions are involved in the phototoxic reactions occurring in the retina, their exact mechanism(s), including the chromophores that photosensitise the photooxidative damage to the retina, are still uncertain.

Figure 7. Light, oxygen and endogenous photosensitizers: all-trans-retinal in photoreceptor outer segments (POS) and lipofuscin in the RPE as factors putting the outer retina - RPE cells and photoreceptors at risk of photooxidative damage.
Retinal chromophores as potential triggers of light damage. The primary requirement for light to exert any photochemical effect is that it needs to be absorbed. In the retina, the incident light is mainly absorbed by visual pigments in photoreceptor outer segments (POS), and by melanin - the most prominent pigment in the young retinal pigment epithelium (RPE) (111). With age there is a gradual accumulation in the RPE of lipofuscin, which reaches substantial concentrations and occupies almost 20% of cytoplasmic volume by the age of 80 years.
However, studies of RPE cells in vitro have demonstrated that these cells are susceptible to blue-light induced damage in the absence of melanin and lipofuscin, while their mitochondria have proved a susceptible target of photodamage (103, 104, 106, 112, 113). Isolated mitochondria have been shown to photogenerate reactive oxygen species, such as singlet oxygen and superoxide, and the action spectrum of singlet oxygen photogeneration is similar to the absorption spectrum of mitochondrial Fe-S centres with the maximum at about 420 nm (106, 114-116). Mitochondrial ubiquinol-cytochrome c reductase has been shown to be the most susceptible enzyme to light-induced inhibition of its activity. Destruction of the Fe-S centres by mersalyl acid substantially decreases the susceptibility of mitochondrial ubiquinol-cytochrome c reductase to photoinduced inhibition. The action spectrum of the remaining photoinhibition can be ascribed to flavins and porphyrins. Potential roles of flavins and porphyrins in retinal photodamage have been previously reviewed in (111). The contributions to photon absorption in the retina by mitochondrial Fe-S centres, flavins and porphyrins, seem to be almost negligible in comparison with contributions from visual pigments, melanin or lipofuscin. Moreover, as will be discussed later, doses of light inducing extensive photodamage to the retina in animals with normal concentrations of visual pigments, are completely safe for animals without visual pigments. However, there is a possibility that chronic photodamage contributes to age-related damage and dysfunction of retinal mitochondria.
Another pigment which accumulates in the primate retina in high concentration is xanthophyll - lutein and zeaxanthin, which are particularly concentrated in the axons of photoreceptors in the fovea (111). Due to a high absorption coefficients corresponding to blue range of the visible spectrum, they act as a blue light filter protecting the outer retina from blue-light-induced injury in the area responsible for acute vision.
Role of visual pigments and vitamin A derivatives in photodamage to the retina. Visual pigments are responsible for visual perception. All visual pigments in vertebrates are formed by a transmembrane protein, opsin, which binds via a Schiff base linkage of its lysine residue to 11-cis-retinal. Differences in amino acid residues in proximity of 11-cis-retinal are responsible for spectral differences in absorption maxima of different types of visual pigments. The visual pigment of rods is named for its colour rhodopsin (from Greek rhodo = rose-red, and opsin = sight) (117). There are three types of cones in human retina, each of which expresses a different visual pigment with absorption maxima at 419 nm (named short-wavelength, S or "blue" cone), 531 nm (middle-wavelength, M or "green" cone) or 558 nm (long-wavelength, L or "red" cone). In contrast to rods, the names of cones refer to the spectral range of light being the most efficiently absorbed in comparison with other cone types (it is important with respect to "red" cone, which exhibits absorption maximum corresponding to yellow light but absorbs red light more efficiently than the other two cone types). Rods are the predominant photoreceptors in human retina, except for a small area in the centre of the fovea, foveola, where only cones are localized.
Presence of visual pigments is an essential factor for light-induced damage to occur. Based on comparison of absorption spectra of visual pigments and wavelength-dependence of photodamage thresholds (action spectra), retinal photodamage can be, at least in part, ascribed to visual pigments of both rods and cones as the chromophores which are initial triggers (94-97, 118, 119). The importance of visual pigments as triggers of light damage in the retina is underscored by experimental findings that animals deficient in rhodopsin are protected from light damage (120). In those experiments, the susceptibility to light damage has been compared between rhodopsin knockout mice (Rho-/-), RPE65 knockout mice (RPE65-/-) and wild type mice. Rho-/- mice are unable to synthesize the apo-protein opsin, and therefore lack rhodopsin, and do not develop photoreceptor outer segments. RPE65-/- mice do not express RPE65 protein essential for synthesis of visual pigment chromophore, 11-cis-retinal, so even though they express the apo-protein opsin, and have morphologically normal retina with photoreceptor outer segments, they lack functional visual pigments. Analysis of the retinal histology 24 h and 7 days after the exposure of dark-adapted animals to 2 h of bright white fluorescent light (15,000 lux) have demonstrated dramatic changes in wild-type animals. The changes were initially evident as disruption and vesiculation of photoreceptor outer and inner segments, and condensation of nuclear chromatin. This was followed by massive degeneration and loss of photoreceptors. In contrast, light exposure did not affect photoreceptors of Rho-/- nor Rpe65-/- mice, which retained retinal morphology similar to the dark-maintained controls. While these experiments have clearly shown that the presence of rhodopsin is the main factor determining the susceptibility to light damage, it needs to be remembered that a retina devoid of rhodopsin is devoid of its primary function - visual perception.
Several other studies also indicate that the degree of retinal photodamage positively correlates with the rhodopsin content in the retina before light exposure (121-123). Availability of dietary all-trans-retinol (Vitamin A) as a precursor of 11-cis-retinal is one of the main determinants of rhodopsin content. Vitamin A deprivation in rats makes them more resistant to photodamage to the retina (121). Synthesis of 11-cis-retinal, and therefore also rhodopsin, can be pharmacologically inhibited due to deficiency of all-trans-retinol (Vitamin A) despite adequate dietary intake. All-trans-retinol normally circulates in the blood plasma and is delivered to the RPE bound to a small 21 kDa monomeric retinoid binding protein (RBP). To prevent glomerular clearance of RBP from blood plasma to urine, RBP binds with a larger 55 kDa tetrameric protein, transthyretin. Depletion of plasma levels of RBP-bound vitamin A can be achieved by an anti-cancer drug Fenretinide [N-(4-hydroxyphenyl) retinamide] (124-126). It has been demonstrated that Fenretinide displaces all-trans-retinol from RBP (126). The complex of Fenretinide with RBP does not bind to transthyretin, so it is rapidly cleared from the plasma. As a result of decreased levels of RBP-all-trans-retinol complex in the plasma, the total content of 11-cis-retinal, and thus also rhodopsin, is substantially reduced in retinas of Fenretinide-treated light-adapted mice, in comparison with vehicle-treated mice.
Another factor affecting rhodopsin expression and its concentration in the newly formed discs of POS is long-term adaptation to environmental light (127, 128). Animals reared under a bright light-dark cycle exhibit lower rhodopsin concentration, and are more resistant to retinal photodamage than animals reared under low intensity light or in the dark (121, 129-131). However, it needs to be mentioned that other adaptive responses, including antioxidant levels and the expression of pro-survivals trophic factors, are also likely to play an important role (129, 130, 132).
The rate of rhodopsin regeneration is an important factor determining the susceptibility of the retina to photodamage. Interestingly, under conditions leading to acute photodamage by bright light, most rhodopsin is bleached within the first few minutes of exposure (99). Subsequent experiments have revealed that the rhodopsin content before exposure to light is not the only determinant of susceptibility to photodamage. The efficiency of rhodopsin regeneration after photobleaching turns out to be another important factor (133, 134) (Figure 8). Slowing down rhodopsin regeneration is an effective way to protect the retina from photodamage (135-137).
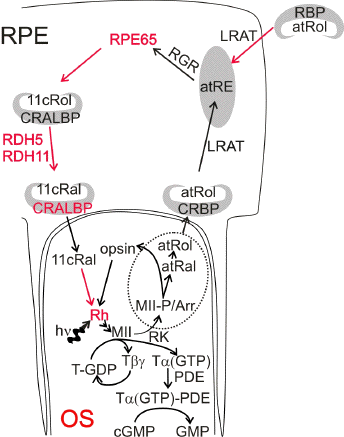
Figure 8. Factors leading to inhibition of rhodopsin regeneration and increased resistance against retinal photodamage in a simplified diagram of phototransduction and visual cycle. Absorption of a photon by rhodopsin (Rh) leads to an ultrafast isomerization of the chromophore, 11-cis-retinal (11cRal) to all-trans-retinal (atRal), which is followed by conformational changes in the protein leading to the formation of a biochemically active state Metarhodopsin II (MII). MII activates a biochemical cascade leading to visual perception: MII binds a heterotrimeric protein transducin (T) and allows for exchange of GDP nucleotide for GTP in a subunitof T. T
and
(T
Slowing down rhodopsin regeneration can be a consequence of certain mutations of RPE65 leading to a diminished isomerase activity of RPE65 protein and therefore limiting the rate of synthesis of 11-cis-retinal (Figure 8) (135, 138-140). A methionine variant of residue 450 of mouse RPE65 (which is normally leucine) is associated with a decreased isomerase activity, inhibition of rhodopsin regeneration and increased resistance of the retina to light damage (138). However, it needs to be remembered that RPE65 mutations in humans leading to inhibition of 11-cis-retinal synthesis also lead to several retinal dystrophies, including Leber's congenital amaurosis, which involve gradual loss of photoreceptors and culminate in blindness at a young age (141-144).
Another protein whose dysfunction leads to inhibition of rhodopsin regeneration is cellular retinal binding protein (CRABP). CRABP normally acts as an acceptor of newly synthesized 11-cis-retinal, and chaperones it within the RPE cells. Albino mice with a non-functional CRABP gene exhibit a 10-fold decreased rate of rhodopsin regeneration and resistance to light-induced damage in comparison with the wild type (145).
Changes in the lipid composition of photoreceptor disc membranes also affect the rate of rhodopsin regeneration (136, 137). Dietary deprivation of docosahexaenoate (DHA) and other omega-3 lipids (which can serve as metabolic precursors of docosahexaenoate) leads to a substantial depletion of DHA in POS discs, reduces the rate of rhodopsin regeneration and prevents light-induced lesions even though the amount of rhodopsin in dark-adapted eyes is increased (136, 137). It needs to be remembered, however, that DHA is extremely important for proper development and function of the retina and other neural tissues, and plays multiple roles as an anti-apoptotic and anti-inflammatory factor (146-148). Thus DHA deprivation is not a useful measure to prevent retinal photodamage.
Another way of inhibiting rhodopsin regeneration is anaesthesia with halothane, which is thought to compete with 11-cis-retinal for the opsin binding site (149-151). Mice and rats anesthetized with halothane are completely protected against retinal injury induced by 60 minute exposure to 13,000 lux of white fluorescent light, whereas animals anesthetized with ketamine and/or xylazine exhibit massive photoreceptor loss induced already by almost 8-fold smaller doses of light (151). Thus, halothane anaesthesia may be considered as a preferable option for ocular surgery during which the retina is likely to be exposed to bright light from an operating microscope.
Another pharmacological approach of slowing down rhodopsin regeneration is delivery of inhibitors of RPE65 and other enzymes involved in synthesis or transport of 11-cis-retinal (152-161). One of well established inhibitors of the retinoid cycle is 13-cis-retinoic acid. 13-cis-retinoic acid is clinically used as a drug called Isotretinoin or Accutane in the treatment of severe acne and certain cancers. Patients taking the drug often develop delayed dark adaptation, which may be perceived as night blindness, a reversible side effect of that treatment. Treatment of experimental rats with 13-cis-retinoic acid has been shown to inhibit RPE65 and 11-cis-retinal dehydrogenase (11cRDH), and effectively protect the retina from light-induced damage (158, 160, 162, 163).
Other inhibitors of the retinoid cycle include 11-fluoro-all-trans-retinol, 11-cis-retinyl bromoacetate, retinylamine and its derivatives, as well as non-retinoid compounds such as farnesylamine and certain isoprenoids (152, 154-157, 159, 164). Experiments on rodents have demonstrated that all-trans-retinylamine is a much more effective inhibitor of the retinoid cycle and exhibits less potential toxicity than other inhibitors, including 13-cis-retinoic acid (152, 156).
The metabolic pathways of retinylamine also suggest that, at least in the short-term, it is a safe retinoid derivative. In the RPE, all-trans-retinylamine is reversibly N-acetylated by lecithin:retinol acyltransferase (LRAT) and stored, like all-trans-retinyl esters, in retinosomes (152, 155). Slow hydrolysis of all-trans-retinylamides in the RPE and liver is believed to provide a supply of all-trans-retinylamine for the long-lasting inhibition of the retinoid cycle. Wild type mice accumulate all-trans-retinylamides in their RPE and liver which remain there for at least a week after a single gavage administration of all-trans-retinylamine (152, 156). Importantly, all-trans-retinylamine does not inhibit LRAT activity with regard to esterification of all-trans-retinol; the rate of esterification of all-trans-retinol is about 50-100 times greater than all-trans-retinylamine (155, 156). The primary action of all-trans-retinylamine is blocking the isomerise activity of RPE65. Thus animals treated with all-trans-retinylamine exhibit increased accumulation of retinyl esters and inhibited synthesis of 11-cis-retinal (152, 155, 156). The inhibitory action is persistent as long as stores of all-trans-retinylamides are available. In LRAT knockout mice, which are unable to synthesize retinylamides, all-trans-retinylamine is rapidly cleared from their eyes and livers, and the rate of 11-cis-retinal production is restored (155). In both wild-type mice and LRAT-/- mice, all-trans-retinylamine can be deaminated to all-trans-retinol, which is then esterified to increase the storage pool of all-trans-retinyl esters. However, in wild-type mice, this pathway of all-trans-retinylamine metabolism is minor in comparison with amide formation.
Notably, pre-treatment of mice with all-trans-retinylamine provides complete protection against light-induced damage to the retina (153). BALB/c mice exposed for 2 h to 5000 lux of white fluorescent light (without pupil dilation) develop within a following week a massive loss of photoreceptors and loss of both scotopic and photopic visual function in the absence of all-trans-retinylamine. Amazingly, mice pre-treated with 3.5
mol all-trans-retinylamine show no significant differences in retinal morphology compared with the dark-maintained control. Based on effectiveness of protection from light-induced retinal injury, all-trans-retinylamine appears to be the most promising retinoid cycle inhibitor for applications in vivo.
Why does the efficiency of rhodopsin regeneration matter in susceptibility of the retina to photodamage? To understand why the efficient regeneration of rhodopsin increases the susceptibility of the retina to photodamage, we need to take a closer look at the retinoid cycle (Figure 8, 9) (133, 134). After absorption of a photon, rhodopsin chromophore, 11-cis-retinal undergoes an ultrafast isomerisation to all-trans-retinal. This primary step is followed by conformational changes of the protein opsin leading to the formation of biochemically active metarhodopsin II. Metarhodopsin II initiates a cascade of events leading to visual perception by activation of G protein, transducin (Figure 8).
Activated transducin activates phosphodiesterase, which then degrades cytosolic cyclic GMP (cGMP) nucleotides. Decreased cytoplasmic levels of cGMP lead to dissociation of cGMP molecules from cGMP-gated channels, which in turn leads to closure of those channels. As a result, the influx of sodium and calcium cations is inhibited, and leads to a build up of positive ions outside the photoreceptor plasma membrane culminating in hyperpolarisation. Metarhodopsin II continues to activate subsequent transducins until it becomes phosphorylated by rhodopsin kinase and binds arrestin. In metarhodopsin II all-trans-retinal remains bound to the lysine, but the Schiff base linkage is deprotonated (Figure 9). Eventually, all-trans-retinal is hydrolysed from the protein but remains non-covalently bound at the opsin "exit site" (165). While remaining in that location, all-trans-retinal can serve as a substrate for an enzyme, photoreceptor retinol dehydrogenase (prRDH), which reduces it to all-trans-retinol. However, if a new molecule of 11-cis-retinal is delivered and bound to the opsin active site, all-trans-retinal dissociates from the protein opsin to the lipid membrane before being enzymatically reduced (165).
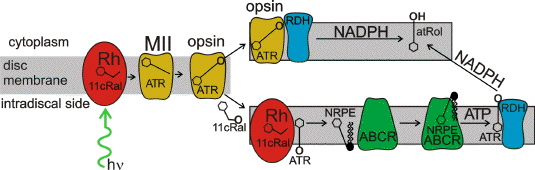
Figure 9. Fate of all-trans-retinal in photoreceptor outer segment disc membrane. Photoactivation of rhodopsin (Rh) leads to formation of metarhodopsin II (MII) from which eventually all-trans-retinal (ATR) is hydrolyzed. There are two pathways leading to enzymatic reduction of all-trans-retinal to all-trans-retinol (atRol) after hydrolysis from opsin. All-trans-retinal can be either reduced by NADPH-dependent photoreceptor dehydrogenase (RDH) while still being bound to the opsin "exit site" or only after it is released to the inner leaflet of the disk membrane as a result of rhodopsin regeneration. Rhodopsin regeneration occurs upon binding to opsin of 11-cis-retinal (11cRal) delivered from the RPE. The reduction of all-trans-retinal is accelerated by ATP-binding cassette transporter rim protein (ABCR, also known as ABCA4), which is present in rims of photoreceptor discs and transports all-trans-retinal complexed with phosphatidylethanolamine (PE), N-retinylidene phophatidylethanolamine (NRPE) to the outer leaflet of disc membrane, where all-trans-retinal can be enzymatically reduced to atRol. Modified from (133, 291).
In mice, the enzymatic reduction all-trans-retinal to all-trans-retinol is a relatively slow process (133, 134). Thus, rhodopsin regeneration is a prerequisite for build-up of free all-trans-retinal in photoreceptor disc membrane. Free all-trans-retinal is not only toxic as a reactive aldehyde and, in the presence of redox active metal ions, a source of free radicals in dark, but it is also a potent photosensitizer photoactivated by UV-A and blue light (133, 134). Photoexcitation of all-trans-retinal with UV-A or blue light is followed by an efficient intersystem crossing from an excited singlet state and formation of an excited triplet state. The energy of retinal triplet state is high enough to enable an efficient energy transfer to molecular oxygen and, as a result, singlet oxygen is produced. The quantum yields of singlet oxygen photogeneration by all-trans-retinal are strongly solvent-dependent. In aprotic, non-polar solvents such as benzene the value of quantum yield of 1O2 generation is 30% (166), whereas in protic methanol - only 5% (167). Photoexcitation of all-trans-retinal in methanol or in dimethylsulphoxide (DMSO):benzene mixture leads to the formation of superoxide radicals (167, 168).
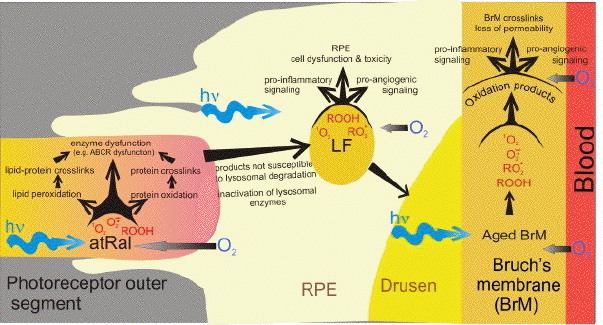
Figure 10. Hypothetical pathways by which all-trans-retinal (atRal) accumulated in photoreceptor outer segments (POS) as a result of photobleaching of visual pigments leads to photodamage of photoreceptors and retinal pigment epithelium (RPE). All-trans-retinal can mediate the generation of superoxide radical anion (O2.-) in the dark, and O2.-, singlet oxygen (1O2), and peroxides (ROOH) when irradiated with UVA or blue light. Unless effective antioxidants and repair enzymes counteract it, reactive oxygen species produced by atRal induce oxidative damage to lipids and proteins, which may affect their structures and functions, including inactivation of enzymes involved in atRal removal (such as ABCR). Tips of the outer segmentsd are phagocytosed daily by the RPE and are meant to undergo lysosomal degradation. However, oxidatively damaged lipids and proteins may no longer be susceptible to degradation by lysosomal enzymes, and/or may inactivate the enzymes. As a result of incomplete lysosomal degradation of the outer segments, residual bodies, called lipofuscin (LF) accumulate in the RPE. LF photoactivated by blue or green light also generates reactive oxygen species and induces further oxidation of intragranular components, some of which may leak out of the granule and cause damage to cellular components of the RPE leading to RPE dysfunction or even death. Some of the oxidation products affect gene expression in the RPE, resulting in a release of pro-inflammatory and pro-angiogenic cytokines. Exocytosed lipofuscin may contribute to the formation of age-related deposits, such as drusen, accumulating between the RPE and Bruch's membrane, which separates the RPE from the choroidal blood supply. Some components of those deposits exhibit photosensitizing properties and include oxidation products with pro-angiogenic and pro-inflammatory properties. Moreover, oxidation leads to formation of crosslinks in the Bruch's membrane contributing to loss of its permeability. Modified from (134).
Both singlet oxygen and superoxide may induce oxidative damage to cellular components (169, 170) (Figure 10). Singlet oxygen directly induces the oxidation of guanine and several amino acid residues, as well as the oxidation of unsaturated lipids, which results in the formation of lipid hydroperoxides. Unsaturated lipids are particularly enriched in the retina, with abundance of docosahexaenoate with six unsaturated double bonds, which is extremely susceptible to peroxidation.
Superoxide can interact with nitric oxide, which is constitutively produced by the retina, and forms extremely reactive peroxynitrite. Peroxynitrite can nitrate lipids and proteins. Indeed, several studies have demonstrated that lipid peroxidation and nitration of proteins is a result of light-induced retinal injury (111, 133, 134). Superoxide dismutates to hydrogen peroxide, which, in turn, may take part in Fenton type reactions: hydrogen peroxide interacts with a reduced form of metal ion such as Cu(I) or Fe(II) leading to oxidation of metal ions and decomposition of the hydrogen peroxide to the hydroxyl anion and extremely oxidizing hydroxyl radical (OH.).
Hydroxyl radicals react rapidly with almost any type of biomolecule, including unsaturated lipids where OH. can initiate a chain of lipid peroxidation. A chain of lipid peroxidation may also be initiated by decomposition of lipid hydroperoxides induced by redox-active metal ions. Secondary products of lipid peroxidation include reactive compounds which add to amino acid residues on proteins, often affecting protein structure and function. Thus, an irradiation of the retina with blue light in the presence of free all-trans-retinal imposes a risk of generation of reactive oxygen species, lipid peroxidation, and oxidative modifications of proteins within photoreceptor outer segments (Figure 10).
It has been shown that photoexcited all-trans-retinal inactivates ATP-binding cassette transporter rim protein, ABCR (also known as ABCA4), which is present in the rims of photoreceptor outer segments discs, and is involved in removal of all-trans-retinal from the discs (171, 172). ABCR functions as a transporter of all-trans-retinal conjugated to phosphatidylethanolamine to the outer leaflet of photoreceptor disc membrane, thus facilitating its enzymatic reduction by retinol dehydrogenase (RDH) (171-176). Inactivation of ABCR may lead to further increase in accumulation of all-trans-retinal as long as there is constant supply of 11-cis-retinal to photobleached visual pigments. Stores of retinyl esters in human RPE account for about 2.5 mol eq of total rhodopsin (177). Thus, in the worst case scenario, where a sufficient supply of 11-cis-retinal is provided, but either ABCR or photoreceptor RDH are inactive, the concentration of accumulated all-trans-retinal in human retina can theoretically reach a staggering concentration of 10.5 to 13 mM (based on rhodopsin concentration of 3 to 3.8 mM) (133, 134).
Free all-trans-retinal forms Schiff base adducts with an abundant component of photoreceptor discs, phosphatidylethanolamine (PE), N-retinylidene phosphatidylethanolamine (NRPE). NRPE may interact with another molecule of all-trans-retinal condensation products forming a bisretinoid, called A2PE (Figure 11). Detection of A2PE and another derivative of all-trans-retinal with PE, all-trans-retinal dimer in the retina, and their corresponding hydrolysis products in the RPE indicate that considerable concentrations of all-trans-retinal do accumulate in the photoreceptor outer segments (133, 134).
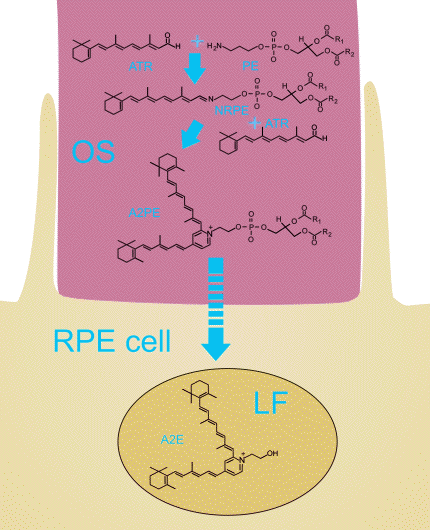
Figure 11. Initial biosynthetic pathway for formation of a lipofuscin fluorophore A2E. All-trans-retinal hydrolysed from opsin binds to a highly abundant outer segment phospholipid, phosphatidylethanolamine (PE), forming N-retinylidene phosphatidylethanolamine (NRPE). Binding of another molecule of all-trans-retinal to NRPE is required to form A2PE. Upon lysosomal cleavage of A2PE in the RPE, A2E is formed and stays there as a component of lipofuscin (LF).
Effects on RPE of photodamage to photoreceptor outer segments. Due to an intimate contact between photoreceptor outer segments (POS) and processes of the RPE surrounding them, it may be suggested that oxidative damage initiated by all-trans-retinal may easily spread from POS to the apical membranes of the RPE. Moreover, the risk of photodamage to the RPE mediated by all-trans-retinal may be increased as POS are constantly renewed and the outer segment tips are phagocytosed daily by the RPE (178). The phagosome fuses with lysosomes, and its content is meant to undergo lysosomal degradation. However, acidification of phagosomes may further intensify the oxidative damage due to protonation of superoxide radical anions, which then become more reactive and capable of initiating a chain of lipid peroxidation. On the other hand, a derivative of all-trans-retinal, a pyridinium bisretinoid called A2E (Figure 11) inhibits lysosomal proton pumps. As a result of an increased pH, activities of lysosomal enzyme are decreased. Certain products of lipid peroxidation have been shown to inhibit lysosomal enzymes directly. Moreover, oxidation of lipids and proteins leads to formation of adducts of proteins with products of lipid oxidation and cross-linked proteins no longer susceptible to degradation by lysosomal enzymes (179). As a result of incomplete lysosomal degradation, residual granular bodies accumulate with age in the RPE called age pigment or lipofuscin (Figure 11) (133, 134).
Lipofuscin accumulation can be significantly reduced by dietary deficiency of vitamin A (ingested as all-trans-retinol, all-trans-retinyl palmitate or its precursors - beta-carotene or cryptoxanthin) or by pharmacological impairment of vitamin A delivery to the eye induced by Fenretinide (126). It has been shown that the rate constants of rhodopsin regeneration after photobleaching are similar in Fenretinide-treated mice and vehicle-treated mice; importantly however, Fenretinide-treated mice exhibit smaller concentrations of accumulated all-trans-retinal after exposure to light.
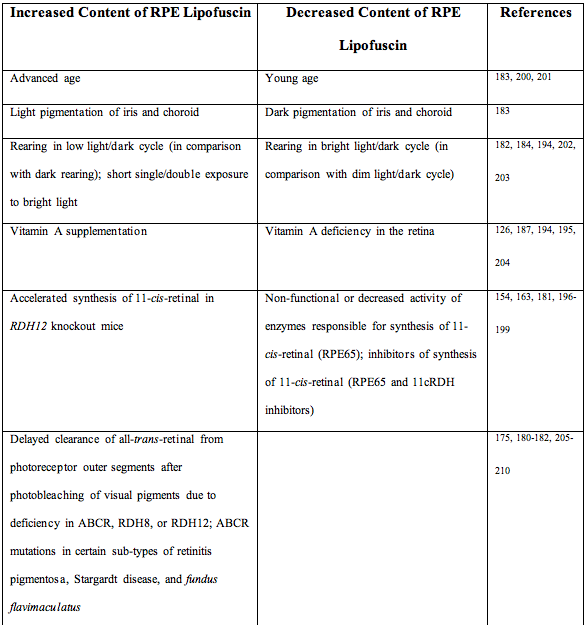
All the conditions discussed earlier that increase susceptibility to retinal photodamage by allowing for efficient regeneration of rhodopsin, and therefore accumulation of all-trans-retinal, have been also shown to accelerate the formation and accumulation of RPE lipofuscin (Table 1) (133, 134). Increased accumulation of lipofuscin is observed in RDH12 knockout mice with an accelerated synthesis of 11-cis-retinal, as well as in ABCR, RDH8, or RDH12 deficient mice in which a delayed clearance of all-trans-retinal from photoreceptor outer segments after photobleaching of visual pigments occurs (175, 180-182). Oxidative stress also increases lipofuscin accumulation (183-193). However, the critical role of all-trans-retinal in formation of RPE lipofuscin is underscored by experiments where rodents receiving intravitreal injections of iron ions accumulate increased lipofuscin in the RPE but lipofuscin accumulation is not increased in animals with vitamin A deficiency despite the injection of iron which does cause severe damage to photoreceptors (187). Consistently, all approaches to inhibit the retinoid cycle and thus minimize accumulation of all-trans-retinal and reduce the risk of photodamage to the retina, also decrease lipofuscin accumulation (126, 154, 163, 187, 194-199) (Table 1). There are also several diseases of human retina and their animal models where increased lipofuscin accumulation is observed and, and while still largely unproven, may be ascribed to an increased accumulation of retinals and oxidative stress in the retina (134) (Table 1).
Table 1. Factors affecting accumulation of lipofuscin in the retinal pigment epithelium (RPE).

Role of lipofuscin in photodamage to the retina. Lipofuscin accumulates progressively throughout life reaching almost 20% of cytoplasmic volume by the age of 80 (200). Due to its broad-band fluorescence when excited with blue or green light, lipofuscin accumulation can be detected not only in histological sections but also in vivo by laser scanning ophthalmoscopy (201, 242, 243).
Accumulation of RPE lipofuscin is greatly accelerated in certain retinal diseases (242). All human diseases related with an increased accumulation of the RPE lipofuscin, as well as some animal models of those diseases, are also related with subsequent dysfunction of the RPE and photoreceptors leading to their atrophy (201, 208, 209, 231, 242, 244, 245). While it still remains to be definitively proven whether lipofuscin is a causal factor in retinal degeneration, there is a growing body of evidence suggesting that lipofuscin can be detrimental to the function and viability of the RPE and neighbouring cells. Age-related accumulation of lipofuscin in whites correlates with loss of underlying photoreceptors (244). The distribution of lipofuscin also correlates with initial degenerative changes observed in age-related macular degeneration (AMD) (201, 246). Measurements of lipofuscin fluorescence and progression of atrophic areas in patients also suggest that areas with increased accumulation of lipofuscin are more likely to become atrophic than other areas (242, 245).
While accumulation of lipofuscin can be a result of photodamage to the retina, once present in the RPE, lipofuscin can itself propagate photo-oxidative damage (133, 134) (Figure 10). With age there is an increase in susceptibility of the RPE to photooxidative damage (133). Several lines of experimental evidence: i) lipofuscin-dependent phototoxicity to cultured human RPE cells; ii) an age-dependent increase in lipofuscin content and susceptibility of RPE cells to photooxidation; and iii) similarities of the action spectra of photo-induced oxidation, indicate that lipofuscin is at least in part responsible (105, 247).
Irradiation of lipofuscin with narrow-band light results in oxygen uptake whose efficiency monotonically increases with decreasing wavelength in the range of 600 nm down to 280 nm (247). Irradiation of lipofuscin granules with blue light leads to photosensitized generation of singlet oxygen which diffuses out of the granule to oxidize extragranular biomolecules (247). Blue light photoexcitation of lipofuscin granules also leads to generation of superoxide, hydrogen peroxide, lipid hydroperoxides and malondialdehyde (247, 248). Hydrogen peroxide accounts for only ~1% of molecular oxygen consumed during the irradiation of lipofuscin, while the majority of oxygen is used for oxidation of intragranular components (249). Lipofuscin contains abundant polyunsaturated lipids, so not surprisingly, irradiation of lipofuscin with visible light leads to the formation of lipid hydroperoxides, and subsequently aldehydic products of lipid peroxidation. Also, extragranular lipids and proteins are susceptible targets of photoinduced oxidation in the presence of photoexcited lipofuscin (247, 249, 250). Chloroform-methanol extraction of lipofuscin gives chloroform soluble lipophilic fraction and chloroform insoluble material (251). Both fractions exhibit substantial photoreactivity.
Photoreactivity of lipophilic components of lipofuscin. Lipophilic extract of lipofuscin exhibits a broad absorption spectrum with absorption coefficient monotonically increasing with decreasing wavelength (Figure 12), and it includes potent photosensitizer(s), which upon photoexcitation form an excited triplet state (166, 252). Lipofuscin triplet state exhibits a broad absorption spectrum with a maximum at about 440 nm and a rate of decay to the ground state of about 1 x 105 s-1 in argon saturated hexane or benzene corresponding to a lifetime of 10.5
s. The triplet interacts with oxygen with a bimolecular rate constant of 1.2 x 109 M-1s-1.
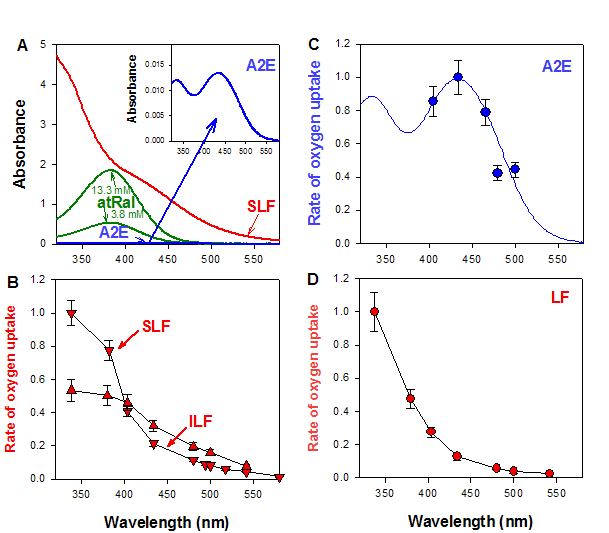
Figure 12. (A) Estimated upper limits for absorption of UV and visible light by all-trans-retinal (atRal) and chloroform soluble components of lipofuscin (SLF) and a component of lipofuscin called A2E in the retina. Absorption spectra of all-trans-retinal correspond to 3.8 mM and 13.3 mM solutions of all-trans-retinal in optical pathlength of 31.2
(B, C, D) Wavelength dependence of initial rates of light-induced oxygen uptake normalized to equal number of incident photons (action spectra of photooxidation) for suspension of lipofuscin granules (LF) (B), insoluble (ILF) and soluble (SLF) components of lipofuscin (C) and A2E in liposomes containing unsaturated lipids as oxidation substrate (D). The maximum in each graph was taken as 100%. Note, the rate of photooxidation increases with decreasing wavelength for LF, SLF, and ILF, while for A2E it exhibits a maximum which matches maximum in its absorption spectrum. Modified from (133, 134).
The energy of the lipofuscin triplet state is high enough to be transferred to molecular oxygen to form singlet oxygen (166). The quantum yields of singlet oxygen generation by photoexcited lipofuscin are dependent on the excitation wavelength, solvent and oxygen concentration. Excitation with 355 nm UV light or blue light (420-440 nm) of a lipophilic extract of lipofuscin solubilised in air-saturated benzene results in the generation of singlet oxygen with a quantum yield of about 8% and 5%, respectively. It suggests that there are different photosensitizers involved and/or contributions of chromophores with different photosensiting properties are different at 355 nm than at 420-440 nm.
Photoexcitation of lipofuscin extract in methanol leads to the formation of an excited triplet state with a lifetime of 7
s (252). The quantum yield of photosensitized formation of singlet oxygen in air-saturated methanol is 5% (the same as for all-trans-retinal) (252, 253).
Saturation of lipofuscin solution in benzene with oxygen leads to a substantial increase in quantum yields of singlet oxygen up to 15% and 9% for excitation with 355 nm and blue light, respectively (166). The increase may be explained by the presence of photosensitizers with triplets with lifetimes shorter than 10.5 s. Alternatively, the increased concentration of oxygen may facilitate intersystem crossing of excited singlet states. It has been demonstrated that lipofuscin includes different fluorophores with singlet state lifetimes of about 60 ps, 0.32 ns, 1.2 ns, and 4.8 ns. Particularly the latter two fluorophores in their excited singlet states are sufficiently long-lived to allow for an efficient interaction with ground state oxygen, leading to an enhanced intersystem crossing to form lipofuscin triplet state and/or an energy transfer from an excited lipofuscin singlet state to oxygen and formation of singlet oxygen.
Superoxide is a minor product generated by photoexcited lipofuscin in comparison with singlet oxygen. The quantum yield of blue light-induced generation of superoxide in a 9:1 mixture of dimethylsulfoxide and benzene is only ~0.1%, that is about 50 times smaller than for singlet oxygen (254).
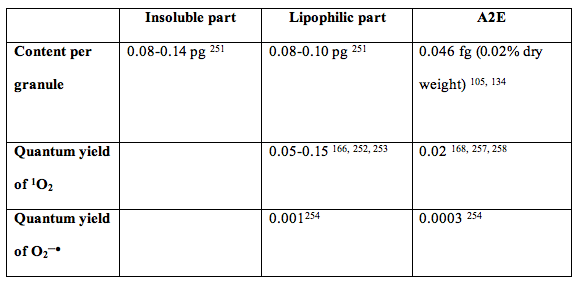
One of lipophilic components of lipofuscin is A2E (255, 256). A2E provides only 0.8% contribution to absorption of visible light by lipofuscin granule, and exhibits very weak photosensitising properties, so the contribution of A2E to the photoreactivity of lipofuscin is only minor (133, 134) (Table 2, Figure 12). For example, A2E contributes at most 1 singlet oxygen molecule per 300 singlet oxygen molecules generated by lipofuscin. A2E contributes at most 1 superoxide molecule per 384 superoxide molecules generated by lipofuscin.
Table 2. Contributions of lipofuscin extracts and one of its component, A2E to content of lipofuscin granule, and comparison of photosensitizing properties of lipophilic extract of lipofuscin with A2E (133, 134).
A2E has attracted a great deal of attention due to its (photo)toxic properties to RPE cells studied in vitro (255). However, studies of lipofuscin phototoxicity demonstrated dramatic phototoxic effects induced by concentrations of lipofuscin corresponding to concentrations of A2E of only 13 ng per million cells, which is at least two orders of magnitude smaller than the A2E concentration needed to exert detectable toxic effects on RPE cells in the dark, and about 17 times smaller than the lowest A2E concentration needed to exert toxicity upon exposure to a 450 J/cm2 dose of blue-green light (390-550 nm) (105, 259). Cells fed with 300 lipofuscin granules per cell and exposed to doses of blue-green light up to 121 J/cm2 exhibit inhibition of antioxidant and lysosomal enzymes, changes in morphology with loss of integrity of the monolayer, loss of lysosomal integrity, enhanced accumulation of lipid peroxidation-derived aldehydes, malondialdehyde and 4-hydroxynonenal, DNA damage and greatly reduced cell viability (105, 106, 260, 261).
Most studies of (photo)toxic effect of A2E were performed with A2E solution in dimethylsulphoxide injected to cell culture medium (133, 134). Under physiological conditions A2E is present in the lipofuscin granule, and recent studies indicate that A2E is strongly anchored within that granule (179). Incubation of lipofuscin granules in the presence of proteinase K and SDS leading to removal of most proteins from the granule, does not affect the concentration of A2E still remaining in the granule. It can be suggested that due to encapsulation of A2E in the lipofuscin granule, A2E is unlikely to exert any deleterious effects on mitochondria, DNA, and transport proteins, which were observed in experiments with delivery of A2E in solution.
Within lipofuscin granules, A2E is very likely to undergo photooxidation. Oxidation products of A2E include a variety of epoxides, cyclic peroxides, furanoid oxides and carbonyls (262-270). Several of these products have been identified in human RPE post mortem. Again, in vitro studies with delivery of A2E to cultured RPE cells in solution and subsequent photooxidation, or direct delivery of A2E oxidation products of A2E in solution have demonstrated several detrimental effects of A2E oxidation products including DNA damage, induction of pro-angiogenic factors, activation of complement cascade and other pro-inflammatory pathways (133, 134). It remains to be shown whether those A2E oxidation products can stimulate those effects while being trapped in the lipofuscin granule.
Another identified lipophilic component of lipofuscin is all-trans-retinal dimer-phosphatidylethanolamine, as well as its derivatives, all-trans-retinal dimer-ethanolamine and all-trans-retinal dimer, and their protonated forms (271). It has been demonstrated that all-trans-retinal dimer and its derivatives generate singlet oxygen upon photoexcitation with 430 nm or 500 nm light, but the yields of singlet oxygen have not been quantified. Interestingly, oxidation products of docosahexaenoate, an abundant component of photoreceptor outer segment membranes and also present in lipofuscin, have been shown to exhibit photosensitizing properties upon photoexcitation with UV or blue light (272). The absorption spectrum of a mixture of oxidized docosahexaenoate exhibits an increasing absorption coefficient with decreasing wavelength in a range of 300-600 nm. Photoexcitation of oxidized docosahexaenoate with 355 nm or blue light leads to the formation of a triplet state similar to that of lipophilic extract of lipofuscin. The triplet state is quenched by oxygen, which leads to photosensitized generation of singlet oxygen. Continuous irradiation of oxidized docosahexaenoate with blue light leads to photosensitized generation of superoxide.
Photoreactivity of chloroform-insoluble components of lipofuscin. Chloroform-insoluble components of lipofuscin also exhibit the ability to photosensitize the generation of singlet oxygen, superoxide and oxidation of exogenous lipids and proteins (251). Both the soluble and insoluble parts of lipofuscin granule undergo photooxidation, and exhibit the ability to oxidize exogenously added lipids and proteins. Interestingly, both soluble and insoluble fractions of lipofuscin demonstrate no age-related changes in photoreactivity when studied at the same concentration of dry mass, even though lipofuscin granules become more photoreactive with age. With age there is an increase in content of insoluble components in an average lipofuscin granule, while the contribution of the soluble part remains constant. Thus, the age-related increase in photoreactivity of an average lipofuscin granule can be explained by the increase of the insoluble part.
Is the photoreactivity of RPE lipofuscin harmful to the retina?
In vivo, lipofuscin granules are continually exposed to visible light (400-700 nm) and high oxygen tensions [about 70 mm Hg (109)], thus providing ideal conditions for the formation of reactive species, with the potential to damage cellular proteins and lipid membranes. However, the retina is equipped with a number of antioxidants and detoxification enzymes. Thus, it may be argued that in order to cause damage, the fluxes of reactive species generated by lipofuscin need to exceed the capacity of those cellular defences.
How is the Retina Protected From Light-Induced Damage?
There are many natural mechanisms protecting the retina from excessive exposure to light. They include ocular geometry where the light is partly blocked by the eyelids from entering the eye through the pupil (273). Aversion response to bright light and squinting further protect from excessive exposure. Furthermore, dilation or constriction of the pupil (known as pupillary light reflex) is responsible for adjusting the light levels reaching the retina within two orders of magnitude.
Long-term exposure to environmental light results in several adaptive responses of the retina. As mentioned earlier, depending on light levels the animals are reared at, the concentration of rhodopsin is regulated, so the resultant flux of absorbed photons in the rod outer segment layer is relatively steady, independent of the seasonal changes of intensity of the environmental light (so called photostasis) (274). For animals reared in bright light rhodopsin synthesis is down-regulated, while ubiquitin-mediated rhodopsin degradation is increased (275, 276). Moreover, rearing animals in bright light results in an increase in the cholesterol content, and a decrease in polyunsaturated lipid content, such as docosahexaenoate, in photoreceptor outer segments (277-280). This change in the lipid environment of rhodopsin in photoreceptor discs strongly decreases the ratio of formation of biochemically active metarhodopsin II to biochemically inactive but thermally stable metarhodopsin III, both of which are formed upon photoexcitation of rhodopsin (133, 134). It can be suggested that as a result of a diminished concentration of metarhodopsin II, the rate of accumulation of free all-trans-retinal is diminished. A substantial depletion of docosahexaenoate in rats distinctly decreases the regeneration rate of rhodopsin and prevents white light-induced retinal injury. These changes in composition in photoreceptor outer segments require time. As it takes about 2 weeks to shed and replace all rod outer segment, it may be argued that this is a minimal time needed to adjust the molecular composition of photoreceptor outer segments to the new intensity of the environmental light. This mechanism is probably sufficient to adapt to seasonal changes in environmental light intensity. However, when we escape for short holidays from a cloudy and rainy country to a place in the sun, or a skiing resort high up in the mountains, we often return back home before our retina becomes fully adapted to the higher light level.
Relatively high concentrations in the retina of low-molecular weight antioxidants, such as hydrophilic ascorbate (vitamin C), or lipophilic alpha-tocopherol (vitamin E) or xanthophylls, lutein and zeaxanthin can protect biomolecules from photosensitized damage by scavenging free radicals, breaking chains of lipid peroxidation and quenching excited triplet states and singlet oxygen (170, 281). The outer retina exhibits also high concentrations of antioxidant enzymes (281, 282). Antioxidant enzymes, such as superoxide dismutase, catalase and glutathione peroxidise are responsible for catalysis of dismutation of superoxide to hydrogen peroxide, and for decomposition of hydrogen peroxide and lipid hydroperoxides (Figure 13).
It is important to stress that there is no single antioxidant that can offer better protection than an appropriate mixture of different antioxidants (Figure 13). For example, lipophilic alpha-tocopherol and hydrophilic ascorbate can offer synergistic protection (170). As a result of scavenging of peroxyl radicals, alpha-tocopherol breaks a chain of lipid peroxidation but becomes a free radical itself. While the alpha-tocopheroxyl radical is much less reactive than lipid-derived peroxyl radicals, its build-up in the lipid membrane may eventually lead to a propagation of lipid peroxidation. Hydrophilic ascorbate can reduce alpha-tocopheroxyl radical back to the parent molecule, thus regenerating the lipophilic antioxidant ready to scavenge another peroxyl radical. As a result of hydrogen transfer, ascorbate becomes a hydrophilic ascorbyl radical. Ascorbyl radicals disproportionate to ascorbate and dehydroascorbate. Ascorbyl radical and dehydroascorbate can be either enzymatically reduced back to ascorbate, or further metabolized and removed to the blood plasma and then to the urine.
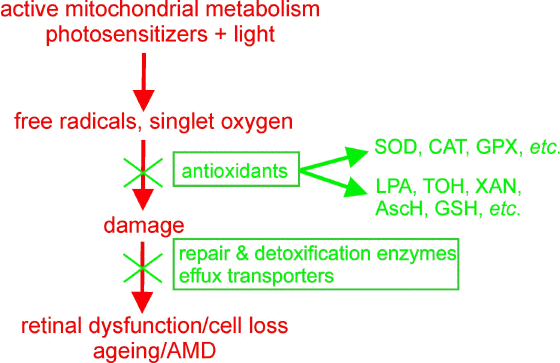
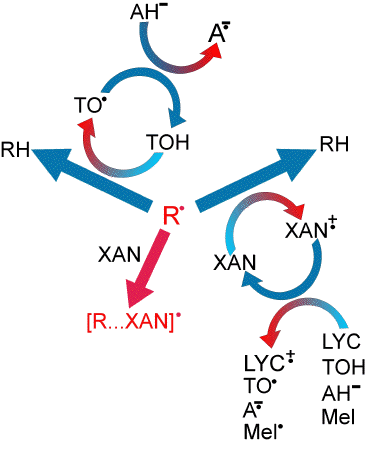
Figure 13. Antioxidant and detoxification pathways potentially involved in protection against photosensitized damage to the retina (Upper Figure). Retinal antioxidants include non-enzymatic antioxidants (such as xanthophylls (XAN), tocopherols (TOH), lipoic acid (LPA), ascorbate (AscH-), glutathione (GSH); enzymatic antioxidants (such as superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidises (GPX); and a number of repair and detoxification enzymes as well as efflux transporters removing damaged biomolecules from the cell. To provide an efficient protection against (photo)oxidative damage a cooperation of different antioxidants is needed.
Lower Figure: An example of mechanisms involved in co-operation of antioxidants. Free radicals (R.) in the lipid membrane can be scavenged by antioxidants, such as lipophilic tocopherol (TOH) or xanthophylls (XAN). XAN can add R. and form a resonance-stabilized radical adduct ([R-XAN].) or reduce R. by electron transfer or hydrogen donation to produce RH. When TOH acts as electron donor, the relatively benign tocopheryl radical (TO.) is formed. The TO. can be reduced, and therefore regenerated to the parent molecule, by ascorbate (AH-). The xanthophyll cation radical (XAN.+) is directly formed when XAN acts as an electron donor. XAN.+ can be recycled to regenerate XAN through a reduction reaction involving lycopene (LYC), TOH, AH- and/or melanin (Mel). The benign lycopene cation radical (LYC.+), TO., ascorbyl radical (A-), and/or melanin radical (Mel.) are formed. Thus, in the presence of other antioxidants, such as TOH, AH- and Mel, XAN can be spared from degradation and therefore may provide longer lived protection as singlet oxygen quenchers.
Another example of cooperation between antioxidants to achieve synergistic protection from photosensitized damage is the cooperation between lipophilic carotenoid, zeaxanthin and either lipophilic alpha-tocopherol or hydrophilic ascorbate (283, 284). Due to its low energy excited triplet state, zeaxanthin is an efficient quencher of excited triplet states of photosensitizers and singlet oxygen. However, it loses those properties when degraded due to interaction with free radicals. Ascorbate and alpha-tocopherol can reduce and therefore regenerate parent xanthophylls from the semi-oxidized xanthophyll cation radical. Moreover, by scavenging free radicals more effectively than xanthophylls, they can protect the xanthophyll from degradation, which allows it to function longer as a quencher of electronically excited states.
When the extent of peroxidation exceeds the capacity of the antioxidant defences, multiple end-products of lipid peroxidation are formed, including reactive carbonyls, and epoxides. As they are often hydrophobic, an efficient detoxification enzyme and transport system has evolved which eliminates these products from the cell (285-288). For example, glutathione transferase can conjugate lipid-derived aldehydes with glutathione, which makes them more water soluble to enable their removal from the lipid membrane, and then from the cell. Long-term adaptation to the environmental light also includes increased concentrations of low-molecular-weight antioxidants in the retina, such as vitamin E and vitamin C, and their levels increase proportionally to the increase in light intensity (279, 280). All these adaptive mechanisms to environmental light are at least partly effective in preventing light-induced damage to the retina (133, 134).
Conclusion
Visible light reaching the retina is essential for visual perception, but, despite the retina being equipped with several mechanisms to protect itself, it is easy to expose the retina to light levels that exceed these natural defences and cause damage. The life-long build-up of oxidative damage, part of which is due to light-induced damage, can contribute to the age-related changes and degenerations observed in the aged retina. A better understanding of the photo-induced processes in the retina is needed to help to predict what levels of illumination are safe for the normal retina, and elucidate the conditions under which even ambient solar radiation may impose a risk of retinal photodamage.
References
1. Boettner EA, Wolter JR. Transmission of the ocular media. Invest Ophthalmol 1962;1:776-783.
2. Jaffe GJ, Irvine AR, Wood IS, Severinghaus JW, Pino GR, Haugen C. Retinal phototoxicity from the operating microscope - the role of inspired oxygen. Ophthalmology 1988;95:1130-1141.
3. Ham WT, Mueller HA, Ruffolo JJ, Millen JE, Cleary SF, Guerry RK, Guerry D. Basic mechanisms underlying the production of photochemical lesions in the mammalian retina. Curr Eye Res 1984;3:165-174.
4. Ruffolo J, Jr, Ham W, Jr, Mueller H, Millen J. Photochemical lesions in the primate retina under conditions of elevated blood oxygen. Invest Ophthalmol Vis Sci 1984;25:893-898.
5. Delori FC, Webb RH, Sliney DH. Maximum permissible exposures for ocular safety (ANSI 2000), with emphasis on ophthalmic devices. J Opt Soc Am A-Opt Image Sci Vis 2007;24:1250-1265.
6. Ozdek S, Deren YT, Gurelik G, Hasanreisoglu B. Posterior subtenon triamcinolone, intravitreal triamcinolone and grid laser photocoagulation for the treatment of macular edema in branch retinal vein occlusion. Ophthalmic Res 2008;40:26-31.
7. Margolis R, Singh RP, Bhatnagar P, Kaiser PK. Intravitreal triamcinolone as adjunctive treatment to laser panretinal photocoagulation for concomitant proliferative diabetic retinopathy and clinically significant macular oedema. Acta Ophthalmol 2008;86:105-110.
8. Misiuk-Hojlo M, Krzyzanowska-Berkowska P, Hill-Bator A. Therapeutic application of lasers in ophthalmology. Adv Clin Exp Med 2007;16:801-805.
9. Meyer CH. Current treatment approaches in diabetic macular edema. Ophthalmologica 2007;221:118-131.
10. Park JJ, Pavesio C. Prophylactic laser photocoagulation for acute retinal necrosis. Does it raise more questions than answers? Br J Ophthalmol 2008;92:1161-1162.
11. Weinstein GW, Rylander HG. Photocoagulation of the fovea. Trans Am Ophthalmol Soc 1978;76:278-295.
12. Friedman E, Kuwabara T. The retinal pigment epithelium. IV. The damaging effects of radiant energy. Arch Ophthalmol 1968;80:265-279.
13. Ham WT, Jr., Ruffolo JJ, Jr., Mueller HA, Clarke AM, Moon ME. Histologic analysis of photochemical lesions produced in rhesus retina by short-wave-length light. Invest Ophthalmol Vis Sci 1978;17:1029-1035.
14. Duke-Elder S, MacFaul PA. Nonmechanical Injuries. In: Duke-Elder S (ed), Systemtis of Ophthalmology. St Louis: CV Mosby; 1972:837-916.
15. Topouzis F, Koskosas A, Pappas T, Anastasopoulos E, Raptou A, Psilas K. Foveomacular retinitis and associated optical coherence tomography findings. Ophthalmic Surg Lasers Imaging 2007;38:333-335.
16. Stangos AN, Petropoulos IK, Pournaras JA, Zaninetti M, Borruat FX, Pournaras CJ. Optical coherence tomography and multifocal electroretinogram findings in chronic solar retinopathy. Am J Ophthalmol 2007;144:131-134.
17. Kallmark FP, Ygge J. Photo-induced foveal injury after viewing a solar eclipse. Acta Ophthalmol Scand 2005;83:586-589.
18. Arda H, Oner A, Mutlu S, Kose Z, Gumus K, Karakucuk S, Mirza E. Multifocal electroretinogram for assessing sun damage following the solar eclipse of 29 March 2006: multifocal electroretinography in solar maculopathy. Doc Ophthalmol 2007;114:159-162.
19. Schatz P, Eriksson U, Ponjavic V, Andreasson S. Multifocal electroretinography and optical coherence tomography in two patients with solar retinopathy. Acta Ophthalmol Scand 2004;82:476-480.
20. Kaushik S, Gupta V, Gupta A. Optical coherence tomography findings in solar retinopathy. Ophthalmic Surg Lasers Imaging 2004;35:52-55.
21. Jorge R, Costa RA, Quirino LS, Paques MW, Calucci D, Cardillo JA, Scott IU. Optical coherence tomography findings in patients with late solar retinopathy. Am J Ophthalmol 2004;137:1139-1143.
22. Garg SJ, Martidis A, Nelson ML, Sivalingam A. Optical coherence tomography of chronic solar retinopathy. Am J Ophthalmol 2004;137:351-354.
23. Ukponmwan CU, Dawodu OA, Ayanru JO. Solar retinopathy in Benin City, Nigeria. West Afr J Med 2003;22:356-357.
24. Doyle E, Sahu D, Ong G. Solar retinopathy after the 1999 solar eclipse in East Sussex. Eye 2002;16:203-206.
25. Codenotti M, Patelli F, Brancato R. OCT findings in patients with retinopathy after watching a solar eclipse. Ophthalmologica 2002;216:463-466.
26. Awan AA, Khan T, Mohammad S, Arif AS. Eclipse retinopathy: follow up of 36 cases after April 1995 solar eclipse in Pakistan. J Ayub Med Coll Abbottabad 2002;14:8-10.
27. Wong SC, Eke T, Ziakas NG. Eclipse burns: a prospective study of solar retinopathy following the 1999 solar eclipse. Lancet 2001;357:199-200.
28. Michaelides M, Rajendram R, Marshall J, Keightley S. Eclipse retinopathy. Eye 2001;15:148-151.
29. Rai N, Thuladar L, Brandt F, Arden GB, Berninger TA. Solar retinopathy. A study from Nepal and from Germany. Doc Ophthalmol 1998;95:99-108.
30. Kawa P, Mankowska A, Mackiewicz J, Zagorski Z. [Solar retinopathy]. Klin Oczna 1998;100:235-237.
31. Atmaca LS, Idil A, Can D. Early and late visual prognosis in solar retinopathy. Graefes Arch Clin Exp Ophthalmol 1995;233:801-804.
32. Hope-Ross MW, Mahon GJ, Gardiner TA, Archer DB. Ultrastructural findings in solar retinopathy. Eye 1993;7 ( Pt 1):29-33.
33. Yannuzzi LA, Fisher YL, Krueger A, Slakter J. Solar retinopathy: a photobiological and geophysical analysis. Trans Am Ophthalmol Soc 1987;85:120-158.
34. Devadason DS, Mahmood S, Stanga PE, Bishop PN. Solar retinopathy in a patient with bipolar affective disorder. Br J Ophthalmol 2006;90:247.
35. Stokkermans TJ, Dunbar MT. Solar retinopathy in a hospital-based primary care clinic. J Am Optom Assoc 1998;69:625-636.
36. Hope-Ross M, Travers S, Mooney D. Solar retinopathy following religious rituals. Br J Ophthalmol 1988;72:931-934.
37. Cangelosi GC, Newsome DA. Solar retinopathy in persons on religious pilgrimage. Am J Ophthalmol 1988;105:95-97.
38. Eigner EH. Self-induced solar retinitis. Am J Ophthalmol 1966;61:1546-1547.
39. Anaclerio AM, Wicker HS. Self-induced solar retinopathy by patients in a psychiatric hospital. Am J Ophthalmol 1970;69:731-736.
40. Freedman J, Gombos GM. Fluorescein fundus angiography in self-induced solar retinopathy. A case report. Can J Ophthalmol 1971;6:124-127.
41. Schatz H, Mendelblatt F. Solar retinopathy from sun-gazing under the influence of LSD. Br J Ophthalmol 1973;57:270-273.
42. Fuller DG. Severe solar maculopathy associated with the use of lysergic acid diethylamide (LSD). Am J Ophthalmol 1976;81:413-416.
43. Gartner J. Long-term follow-up of an ophthalmologist's central serous retinopathy, photocoagulated by sungazing. Doc Ophthalmol 1987;66:19-33.
44. Sadun AC, Sadun AA, Sadun LA. Solar retinopathy. A biophysical analysis. Arch Ophthalmol 1984;102:1510-1512.
45. Gladstone GJ, Tasman W. Solar retinitis after minimal exposure. Arch Ophthalmol 1978;96:1368-1369.
46. van de Kraats J, van Norren D. Optical density of the aging human ocular media in the visible and the UV. J Opt Soc Am A-Opt Image Sci Vis 2007;24:1842-1857.
47. Tso MO, La Piana FG. The human fovea after sungazing. Trans Sect Ophthalmol Am Acad Ophthalmol Otolaryngol 1974;79:788-795.
48. Green WR, Robertson DM. Pathologic findings of photic retinopathy in the human eye. Am J Ophthalmol 1991;112:520-527.
49. Rothkoff L, Kushelevsky A, Blumenthal M. Solar retinopathy: visual prognosis in 20 cases. Isr J Med Sci 1978;14:238-243.
50. Ham WT, Mueller HA, Ruffolo JJ, Clarke AM. Sensitivity of the retina to radiation damage as a function of wavelength. Photochem Photobiol 1978;29:735-743.
51. Kraushar MF. Foveal diseases. Ann Ophthalmol 1986;18:354-357.
52. Kirkness CM. Do ophthalmic instruments pose a hazard of light-induced damage to the eye? In: Cronly-Dillon J, Rosen ES, Marshall J (eds), Hazards of Light; Myths & Realities; Eye and Skin. Oxford: Pergamon Press; 1986:179-186.
53. Lerman S. Effects of sunlight on the eye. In: Ben Hur E, Rosenthal I (eds), Photomedicine. Boca Raton: CRC Press; 1987:79-121.
54. Sliney DH. Optical radiation safety of medical light sources. Phys Med Biol 1997;42:981-996.
55. Michael R, Wegener A. Estimation of safe exposure time from an ophthalmic operating microscope with regard to ultraviolet radiation and blue-light hazards to the eye. J Opt Soc Am A-Opt Image Sci Vis 2004;21:1388-1392.
56. Komaromy AM, Acland GM, Aguirre GD. Operating in the dark: a night-vision system for surgery in retinas susceptible to light damage. Arch Ophthalmol 2008;126:714-717.
57. Parver LM, Auker CR, Fine BS. Observations on monkey eyes exposed to light from an operating microscope. Ophthalmology 1983;90:964-972.
58. Costagliola C, Menzione M, Chiosi F, Romano MR, Della Corte M, Rinaldi M. Retinal phototoxicity induced by hydrochlorothiazide after exposure to a UV tanning device. Photochem Photobiol 2008;84:1294-1297.
59. Barkana Y, Belkin M. Laser eye injuries. Surv Ophthalmol 2000;44:459-478.
60. Naidoff MA, Sliney DH. Retinal injury from a welding arc. Am J Ophthalmol 1974;77:663-668.
61. Romanchuk KG, Pollak V, Schneider RJ. Retinal burn from a welding arc. Can J Ophthalmol 1978;13:120-122.
62. Gardner TW, Ai E, Chrobak M, Shoch DE. Photic maculopathy secondary to short-circuiting of a high-tension electric current. Ophthalmology 1982;89:865-868.
63. Vojnikovic B, Njiric S, Coklo M, Spanjol J. Ultraviolet sun radiation and incidence of age-related macular degeneration on Croatian Island Rab. Coll Antropol 2007;31 Suppl 1:43-44.
64. Plestina-Borjan I, Klinger-Lasic M. Long-term exposure to solar ultraviolet radiation as a risk factor for age-related macular degeneration. Coll Antropol 2007;31 Suppl 1:33-38.
65. Loeffler KU, Sastry SM, McLean IW. Is age-related macular degeneration associated with pinguecula or scleral plaque formation? Curr Eye Res 2001;23:33-37.
66. Young RW. Solar radiation and age-related macular degeneration. Surv Ophthalmol 1988;32:252-269.
67. Taylor HR, West S, Munoz B, Rosenthal FS, Bressler SB, Bressler NM. The long-term effects of visible-light on the eye. Arch Ophthalmol 1992;110:99-104.
68. Tomany SC, Cruickshanks KJ, Klein R, Klein BEK, Knudtson MD. Sunlight and the 10-year incidence of age-related maculopathy - The Beaver Dam Eye Study. Arch Ophthalmol 2004;122:750-757.
69. Delcourt C, Carriere I, Ponton-Sanchez A, Fourrey S, Lacroux A, Papoz L. Light exposure and the risk of age-related macular degeneration: the Pathologies Oculaires Liees a l'Age (POLA) study. Arch Ophthalmol 2001;119:1463-1468.
70. Richer SP. Is there a prevention and treatment strategy for macular degeneration? J Am Optom Assoc 1993;64:838-850.
71. Glazer-Hockstein C, Dunaief JL. Could blue light-blocking lenses decrease the risk of age-related macular degeneration? Retina 2006;26:1-4.
72. LaVail MM, Gorrin GM, Repaci MA, Thomas LA, Ginsberg HM. Genetic regulation of light damage to photoreceptors. Invest Ophthalmol Vis Sci 1987;28:1043-1048.
73. Iseli HP, Wenzel A, Hafezi F, Reme CE, Grimm C. Light damage susceptibility and RPE65 in rats. Exp Eye Res 2002;75:407-413.
74. Danciger M, Lyon J, Worrill D, Hoffman S, Lem J, Reme CE, Wenzel A, Grimm C. New retinal light damage QTL in mice with the light-sensitive RPE65 LEU variant. Mammalian Genome 2004;15:277-283.
75. Paskowitz DM, LaVail MM, Duncan JL. Light and inherited retinal degeneration. Br J Ophthalmol 2006;90:1060-1066.
76. Danciger M, Ogando D, Yang HD, Matthes MT, Yu N, Ahern K, Yasumura D, Williams RW, LaVail MM. Genetic modifiers of retinal degeneration in the rd3 mouse. Invest Ophthalmol Vis Sci 2008;49:2863-2869.
77. Nir I, Liu C, Wen R. Light Treatment Enhances Photoreceptor Survival in Dystrophic Retinas of Royal College of Surgeons Rats. Invest Ophthalmol Vis Sci 1999;40:2383-2390.
78. Organisciak DT, Li M, Darrow RM, Farber DB. Photoreceptor cell damage by light in young Royal College of Surgeons rats. Curr Eye Res 1999;19:188-196.
79. Chrysostomou V, Stone J, Stowe S, Barnett NL, Valter K. The Status of Cones in the Rhodopsin Mutant P23H-3 Retina: Light-Regulated Damage and Repair in Parallel with Rods. Invest Ophthalmol Vis Sci 2008;49:1116-1125.
80. Tam BM, Moritz OL. Dark rearing rescues P23H rhodopsin-induced retinal degeneration in a transgenic Xenopus laevis model of retinitis pigmentosa: A chromophore-dependent mechanism characterized by production of N-terminally truncated mutant rhodopsin. J Neurosci 2007;27:9043-9053.
81. White DA, Hauswirth WW, Kaushal S, Lewin AS. Increased Sensitivity to Light-Induced Damage in a Mouse Model of Autosomal Dominant Retinal Disease. Invest Ophthalmol Vis Sci 2007;48:1942-1951.
82. Cideciyan AV, Jacobson SG, Aleman TS, Gu D, Pearce-Kelling SE, Sumaroka A, Acland GM, Aguirre GD. In vivo dynamics of retinal injury and repair in the rhodopsin mutant dog model of human retinitis pigmentosa. Proc Natl Acad Sci U S A 2005;102:5233-5238.
83. Organisciak DT, Darrow RM, Barsalou L, Kutty RK, Wiggert B. Susceptibility to Retinal Light Damage in Transgenic Rats with Rhodopsin Mutations. Invest Ophthalmol Vis Sci 2003;44:486-492.
84. Vaughan DK, Coulibaly SF, Darrow RM, Organisciak DT. A Morphometric Study of Light-Induced Damage in Transgenic Rat Models of Retinitis Pigmentosa. Invest Ophthalmol Vis Sci 2003;44:848-855.
85. Wang M, Lam TT, Tso MO, Naash MI. Expression of a mutant opsin gene increases the susceptibility of the retina to light damage. Vis Neurosci 1997;14:55-62.
86. Naash ML, Peachey NS, Li ZY, Gryczan CC, Goto Y, Blanks J, Milam AH, Ripps H. Light-induced acceleration of photoreceptor degeneration in transgenic mice expressing mutant rhodopsin. Invest Ophthalmol Vis Sci 1996;37:775-782.
87. Cremers FP, Maugeri A, den Hollander AI, Hoyng CB. The expanding roles of ABCA4 and CRB1 in inherited blindness. Novartis Found Symp 2004;255:68-79; discussion 79-84, 177-178.
88. Berson EL. Light deprivation for early retininitis pigmentosa - Hypothesis. Arch Ophthalmol 1971;85:521-529.
89. Stone J, Maslim J, Valter-Kocsi K, Mervin K, Bowers F, Chu Y, Barnett N, Provis J, Lewis G, Fisher SK, Bisti S, Gargini C, Cervetto L, Merin S, Pe'er J. Mechanisms of photoreceptor death and survival in mammalian retina. Prog Retin Eye Res 1999;18:689-735.
90. Heckenlively JR, Rodriguez JA, Daiger SP. Autosomal dominant sectoral retinitis pigmentosa. Two families with transversion mutation in codon 23 of rhodopsin. Arch Ophthalmol 1991;109:84-91.
91. Organisciak DT, Winkler BS. Retinal light damage: practical and theoretical considerations. Prog Retin Eye Res 1994;13:1-29.
92. Wenzel A, Grimm C, Samardzija M, Reme CE. Molecular mechanisms of light-induced photoreceptor apoptosis and neuroprotection for retinal degeneration. Prog Retin Eye Res 2005;24:275-306.
93. Marc RE, Jones BW, Watt CB, Vazquez-Chona F, Vaughan DK, Organisciak DT. Extreme retinal remodeling triggered by light damage: implications for age related macular degeneration. Mol Vis 2008;14:782-806.
94. Noell WK, Walker VS, Kang BS, Berman S. Retinal damage by light in rats. Invest Ophthalmol 1966;5:450-473.
95. Williams TP, Howell WL. Action spectrum of retinal light-damage in albino rats. Invest Ophthalmol Vis Sci 1983;24:285-287.
96. Harwerth RS, Sperling HG. Effects of intense visible radiation on the increment-threshold spectral sensitivity of the rhesus monkey eye. Vision Res 1975;15:1193-1204.
97. Sperling HG, Johnson C, Harwerth RS. Differential spectral photic damage to primate cones. Vision Res 1980;20:1117-1125.
98. Ham WT, Mueller HA, Ruffolo JJ, Guerry D, Guerry RK. Action spectrum for retinal injury from near-ultraviolet radiation in the aphakic monkey. Am J Ophthalmol 1982;93:299-306.
99. Gorgels TGMF, van Norren D. Ultraviolet and green light cause different types of damage in rat retina. Invest Ophthalmol Vis Sci 1995;36:851-863.
100. Bush EM, Gorgels TGMF, van Norren D. Temporal sequence of changes in rat retina after UV-A and blue light exposure. Vision Res 1999;39:1233-1247.
101. Tso MO, Fine BS. Repair and late degeneration of the primate foveola after injury by argon laser. Invest Ophthalmol Vis Sci 1979;18:447-461.
102. Tso MO. Experiments on visual cells by nature and man: in search of treatment for photoreceptor degeneration. Friedenwald lecture. Invest Ophthalmol Vis Sci 1989;30:2430-2454.
103. Dorey CK, Delori FC, Akeo K. Growth of cultured RPE and endothelial cells is inhibited by blue light but not green or red light. Curr Eye Res 1990;9:549-559.
104. Pautler EL, Morita M, Beezley D. Reversible and irreversible blue light damage to the isolated, mammalian pigment epithelium. Prog Clin Biol Res 1989;314:555-567.
105. Davies S, Elliott MH, Floor E, Truscot TG, Zareba M, Sarna T, Shamsi FA, Boulton ME. Photocytotoxicity of lipofuscin in human retinal pigment epithelial cells. Free Radic Biol Med 2001;31:256-265.
106. Godley BF, Shamsi FA, Liang FQ, Jarrett SG, Davies S, Boulton M. Blue light induces mitochondrial DNA damage and free radical production in epithelial cells. J Biol Chem 2005;280:21061-21066.
107. Crockett RS, Lawwill T. Oxygen dependence of damage by 435 nm light in cultured retinal epithelium. Curr Eye Res 1984;3:209-215.
108. DeLint PJ, VanNorren D, Toebosch AMW. Effect of body temperature on threshold for retinal light damage. Invest Ophthalmol Vis Sci 1992;33:2382-2387.
109. Ahmed J, Braun RD, Dunn R, Jr., Linsenmeier RA. Oxygen distribution in the macaque retina. Invest Ophthalmol Vis Sci 1993;34:516-521.
110. Linsenmeier RA. Effects of light and darkness on oxygen distribution and consumption in the cat retina. J Gen Physiol 1986;88:521-542.
111. Boulton M, Rozanowska M, Rozanowski B. Retinal photodamage. J Photochem Photobiol B-Biol 2001;64:144-161.
112. Pang JJ, Seko Y, Tokoro T. Processes of pine light-induced damage to retinal pigment epithelial cells lacking phagosomes. Jpn J Ophthalmol 1999;43:103-108.
113. Seko Y, Pang JJ, Tokoro T, Ichinose S, Mochizuki M. Blue light-induced apoptosis in cultured retinal pigment epithelium cells of the rat. Graefes Arch Clin Exp Ophthalmol 2001;239:47-52.
114. Jung J, Kim YJ. Inactivation of citric acid cycle enzymes as a result of photodynamic sensitizarion by mitochondrial inner membrane. Photochem Photobiol 1990;52:1011-1015.
115. Kim CS, Jung J. Iron sulfur centers as endogenous blue-light sensitizers in cells - a study with an artificial nonheme iron protein. Photochem Photobiol 1992;56:63-68.
116. Kim CS, Jung J. Inactivation of the respiratory chain in plant mitochondria by visible light - the primary target for photodamage and endogenous photosensitizing chromophores. J Photochem Photobiol B-Biol 1995;29:135-139.
117. Rodieck RW. The First Steps in Seeing. Sunderland, Massachusetts: Sinauer Associates, Inc.; 1998.
118. Kuwabara T, Gorn RA. Retinal damage by visible light. An electron microscopic study. Arch Ophthalmol 1968;79:69-78.
119. Gorn RA, Kuwabara T. Retinal damage by visible light. A physiologic study. Arch Ophthalmol 1967;77:115-118.
120. Grimm C, Wenzel A, Hafezi F, Yu S, Redmond TM, Reme CE. Protection of RPE65-deficient mice identifies rhodopsin as a mediator of light-induced retinal degeneration. Nature Genetics 2000;25:63-66.
121. Noell WK. Effects of environmental lighting and dietary vitamin A on the vulnerability of the retina to light damage. Photochem Photobiol 1979;29:717-723.
122. Organisciak DT, Noell WK. The rod outer segment phospholipid/opsin ratio of rats maintained in darkness or cyclic light. Invest Ophthalmol Vis Sci 1977;16:188-190.
123. Noell WK, Albrecht R. Irreversible effects on visible light on the retina: role of vitamin A. Science 1971;172:76-79.
124. Caruso RC, Zujewski J, Iwata F, Podgor MJ, Conley BA, Ayres LM, Kaiser-Kupfer MI. Effects of Fenretinide (4-HPR) on dark adaptation. Arch Ophthalmol 1998;116:759-763.
125. Baglietto L, Torrisi R, Arena G, Tosetti F, Gonzaga AG, Pasquetti W, Robertson C, Decensi A. Ocular effects of fenretinide, a vitamin A analog, in a chemoprevention trial of bladder cancer. Cancer Detect Prev 2000;24:369-375.
126. Radu RA, Han Y, Bui TV, Nusinowitz S, Bok D, Lichter J, Widder K, Travis GH, Mata NL. Reductions in serum vitamin A arrest accumulation of toxic retinal fluorophores: A potential therapy for treatment of lipofuscin-based retinal diseases. Invest Ophthalmol Vis Sci 2005;46:4393-4401.
127. Williams TP, Squitieri A, Henderson RP, Webbers JPP. Reciprocity between light intensity and rhodopsin concentration across the rat retina. J Physiol 1999;516:869-874.
128. Organisciak DT, Xie A, Wang HM, Jiang YL, Darrow RM, Donoso LA. Adaptive changes in visual cell transduction protein levels: effect of light. Exp Eye Res 1991;53:773-779.
129. Kaldi I, Martin RE, Huang H, Brush RS, Morrison KA, Anderson RE. Bright cyclic rearing protects albino mouse retina against acute light-induced apoptosis. Mol Vis 2003;9:337-344.
130. Li F, Cao W, Anderson RE. Alleviation of constant-light-induced photoreceptor degeneration by adaptation of adult albino rat to bright cyclic light. Invest Ophthalmol Vis Sci 2003;44:4968-4975.
131. Organisciak DT, Darrow RM, Barsalou L, Darrow RA, Kutty RK, Kutty G, Wiggert B. Light history and age-related changes in retinal light damage. Invest Ophthalmol Vis Sci 1998;39:1107-1116.
132. Li F, Cao W, Anderson RE. Protection of photoreceptor cells in adult rats from light- induced degeneration by adaptation to bright cyclic light. Exp Eye Res 2001;73:569-577.
133. Rozanowska M, Sarna T. Light-induced damage to the retina: Role of rhodopsin chromophore revisited. Photochem Photobiol 2005;81:1305-1330.
134. Rozanowska M, Rozanowski B. Visual Transduction and Age-Related Changes in Lipofuscin. In: Tombran-Tink J, Barnstable CJ (eds), Ophthalmology Research: The Visual Transduction Cascade. Totowa, NJ: The Humana Press Inc.; 2008:405-446.
135. Wenzel A, Reme CE, Williams TP, Hafezi F, Grimm C. The RPE65 Leu450Met variation increases retinal resistance against light-induced degeneration by slowing rhodopsin regeneration. J Neurosci 2001;21:53-58.
136. Bush RA, Malnoe A, Reme CE, Williams TP. Dietary deficiency of N-3 fatty acids alters rhodopsin content and function in the rat retina. Invest Ophthalmol Vis Sci 1994;35:91-100.
137. Bush RA, Reme CE, Malnoe A. Light damage in the rat retina - the effect of dietary deprivation of N-3 fatty-acids on acute structural alterations. Exp Eye Res 1991;53:741-752.
138. Redmond TM, Weber CH, Poliakov E, Yu S, Gentleman S. Effect of Leu/Met variation at residue 450 on isomerase activity and protein expression of RPE65 and its modulation by variation at other residues. Mol Vis 2007;13:1813-1821.
139. Nusinowitz S, Nguyen L, Radu R, Kashani Z, Farber D, Danciger M. Electroretinographic evidence for altered phototransduction gain and slowed recovery from photobleaches in albino mice with a MET450 variant in RPE65. Exp Eye Res 2003;77:627-638.
140. Wenzel A, Grimm C, Samardzija M, Reme CE. The genetic modifier RPE65Leu(450): Effect on light damage susceptibility in c-Fos-deficient mice. Invest Ophthalmol Vis Sci 2003;44:2798-2802.
141. Samardzija M, von Lintig J, Tanimoto N, Oberhauser V, Thiersch M, Reme CE, Seeliger M, Grimm C, Wenzel A. R91W mutation in Rpe65 leads to milder early-onset retinal dystrophy due to the generation of low levels of 11-cis-retinal. Hum Mol Genet 2008;17:281-292.
142. Lorenz B, Poliakov E, Schambeck M, Friedburg C, Preising MN, Redmond TM. A novel RPE65 hypomorph expands the clinical phenotype of RPE65 mutations. A comprehensive clinical and biochemical functional study. Invest Ophthalmol Vis Sci 2008.
143. Thompson DA, Gyurus P, Fleischer LL, Bingham EL, McHenry CL, Apfelstedt-Sylla E, Zrenner E, Lorenz B, Richards JE, Jacobson SG, Sieving PA, Gal A. Genetics and phenotypes of RPE65 mutations in inherited retinal degeneration. Invest Ophthalmol Vis Sci 2000;41:4293-4299.
144. den Hollander AI, Roepman R, Koenekoop RK, Cremers FPM. Leber congenital amaurosis: Genes, proteins and disease mechanisms. Prog Retin Eye Res 2008;27:391-419.
145. Saari JC, Nawrot M, Kennedy BN, Garwin GG, Hurley JB, Huang J, Possin DE, Crabb JW. Visual cycle impairment in cellular retinaldehyde binding protein (CRALBP) knockout mice results in delayed dark adaptation. Neuron 2001;29:739-748.
146. Bazan NG. Omega-3 fatty acids, pro-inflammatory signaling and neuroprotection. Curr Opin Clin Nutr Metab Care 2007;10:136-141.
147. Bazan NG. Neurotrophins induce neuroprotective signaling in the retinal pigment epithelial cell by activating the synthesis of the anti-inflammatory and anti-apoptotic neuroprotectin D1. Adv Exp Med Biol 2008;613:39-44.
148. SanGiovanni JP, Chew EY. The role of omega-3 long-chain polyunsaturated fatty acids in health and disease of the retina. Prog Retin Eye Res 2005;24:87-138.
149. Ishizawa Y, Pidikiti R, Liebman PA, Eckenhoff RG. G protein-coupled receptors as direct targets of inhaled anesthetics. Mol Pharmacol 2002;61:945-952.
150. Grimm C, Wenzel A, Williams TP, Rol PO, Hafezi F, Reme CE. Rhodopsin-mediated blue-light damage to the rat retina: Effect of photoreversal of bleaching. Invest Ophthalmol Vis Sci 2001;42:497-505.
151. Keller C, Grimm C, Wenzel A, Hafezi F, Reme CE. Protective effect of halothane anesthesia on retinal light damage: inhibition of metabolic rhodopsin regeneration. Invest Ophthalmol Vis Sci 2001;42:476-480.
152. Golczak M, Maeda A, Bereta G, Maeda T, Kiser PD, Hunzelmann S, von Lintig J, Blaner WS, Palczewski K. Metabolic basis of visual cycle inhibition by retinoid and nonretinoid compounds in the vertebrate retina. J Biol Chem 2008;283:9543-9554.
153. Maeda A, Maeda T, Golczak M, Imanishi Y, Leahy P, Kubota R, Palczewski K. Effects of potent inhibitors of the retinoid cycle on visual function and photoreceptor protection from light damage in mice. Mol Pharmacol 2006;70:1220-1229.
154. Maiti P, Kong J, Kim SR, Sparrow JR, Allikmets R, Rando RR. Small molecule RPE65 antagonists limit the visual cycle and prevent lipofuscin formation. Biochemistry 2006;45:852-860.
155. Golczak M, Imanishi Y, Kuksa V, Maeda T, Kubota R, Palczewski K. Lecithin:retinol acyltransferase is responsible for amidation of retinylamine, a potent inhibitor of the retinoid cycle. J Biol Chem 2005;280:42263-42273.
156. Golczak M, Kuksa V, Maeda T, Moise AR, Palczewski K. Positively charged retinoids are potent and selective inhibitors of the trans-cis isomerization in the retinoid (visual) cycle. Proc Natl Acad Sci U S A 2005;102:8162-8167.
157. Fishkin N, Yefidoff R, Gollipalli DR, Rando RR. On the mechanism of isomerization of all-trans-retinol esters to 11-cis-retinol in retinal pigment epithelial cells: 11-fluoro-all-trans-retinol as substrate/inhibitor in the visual cycle. Bioorg Med Chem 2005;13:5189-5194.
158. Gollapalli DR, Rando RR. The specific binding of retinoic acid to RPE65 and approaches to the treatment of macular degeneration. Proc Natl Acad Sci U S A 2004;101:10030-10035.
159. Gollapalli DR, Rando RR. Specific inactivation of isomerohydrolase activity by 11-cis-retinoids. Biochim Biophys Acta 2003;1651:93-101.
160. Gamble MV, Mata NL, Tsin AT, Mertz JR, Blaner WS. Substrate specificities and 13-cis-retinoic acid inhibition of human, mouse and bovine cis-retinol dehydrogenases. Biochim Biophys Acta 2000;1476:3-8.
161. Law WC, Rando RR. The molecular basis of retinoic acid induced night blindness. Biochem Biophys Res Commun 1989;161:825-829.
162. Sieving PA, Chaudhry P, Kondo M, Provenzano M, Wu D, Carlson TJ, Bush RA, Thompson DA. Inhibition of the visual cycle in vivo by 13-ccis retinoic acid protects from light damage and provides a mechanism for night blindness in isotretinoin therapy. Proc Natl Acad Sci U S A 2001;98:1835-1840.
163. Radu RA, Mata NL, Nusinowitz S, Liu XR, Sieving PA, Travis GH. Treatment with isotretinoin inhibits lipofuscin accumulation in a mouse model of recessive Stargardt's macular degeneration. Proc Natl Acad Sci U S A 2003;100:4742-4747.
164. Maiti P, Kong J, Kim SR, Sparrow JR, Allikmets R, Rando RR. Erratum: Small molecule RPE65 antagonists limit the visual cycle and prevent lipofuscin formation. Biochemistry 2007;46:8700.
165. Schadel SA, Heck M, Maretzki D, Filipek S, Teller DC, Palczewski K, Hofmann KP. Ligand channeling within a G-protein-coupled receptor - The entry and exit of retinals in native opsin. J Biol Chem 2003;278:24896-24903.
166. Rozanowska M, Wessels J, Boulton M, Burke JM, Rodgers MAJ, Truscott TG, Sarna T. Blue light-induced singlet oxygen generation by retinal lipofuscin in non-polar media. Free Radic Biol Med 1998;24:1107-1112.
167. Dillon J, Gaillard ER, Bilski P, Chignell CF, Reszka KJ. The photochemistry of the retinoids as studied by steady-state and pulsed methods. Photochem Photobiol 1996;63:680-685.
168. Pawlak A, Wrona M, Rozanowska M, Zareba M, Lamb LE, Roberts J, Simon JD, Sarna T. Comparison of the aerobic photoreactivity of A2E with its precursor retinal. Photochem Photobiol 2003;77:253-258.
169. Foote CS. Singlet oxygen. In: Pryor WA (ed), Free Radicals in Biology. New York: Academic Press; 1976.
170. Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. 3rd ed. Oxford: Oxford University Press; 2000.
171. Sun H, Nathans J. Mechanistic studies of ABCR, the ABC transporter in photoreceptor outer segments responsible for autosomal recessive Stargardt disease. J Bioenerg Biomembr 2001;33:523-530.
172. Sun H, Nathans J. ABCR, the ATP-binding cassette transporter responsible for Stargardt macular dystrophy, is an efficient target of all-trans-retinal-mediated photooxidative damage in vitro - Implications for retinal disease. J Biol Chem 2001;276:11766-11774.
173. Molday RS, Beharry S, Ahn JH, Zhong M. Binding of N-retinylidene-Pe to ABCA4 and a model for its transport across membranes. Retinal Degenerative Diseases. Berlin: Springer-Verlag Berlin; 2006:465-470.
174. Beharry S, Zhong M, Molday RS. N-retinylidene-phosphatidylethanolamine is the preferred retinoid substrate for the photoreceptor-specific ABC transporter ABCA4 (ABCR). J Biol Chem 2004;279:53972-53979.
175. Weng J, Mata NL, Azarian SM, Tzekov RT, Birch DG, Travis GH. Insights into the function of Rim protein in photoreceptors and etiology of Stargardt's disease from the phenotype in abcr knockout mice. Cell 1999;98:13-23.
176. Ahn J, Wong JT, Molday RS. The effect of lipid environment and retinoids on the ATPase activity of ABCR, the photoreceptor ABC transporter responsible for Stargardt macular dystrophy. J Biol Chem 2000;275:20399-20405.
177. Bridges CDB, Alvarez RA, Fong SL. Vitamin A in human eyes - amount, distribution, and composition. Invest Ophthalmol Vis Sci 1982;22:706-714.
178. Strauss O. The retinal pigment epithelium in visual function. Physiol Rev 2005;85:845-881.
179. Ng K-P, Gugiu B, Renganathan K, Davies MW, Gu X, Crabb JS, Kim SR, Rozanowska MB, Bonilha VL, Rayborn ME, Salomon RG, Sparrow JR, Boulton ME, Hollyfield JG, Crabb JW. Retinal pigment epithelium lipofuscin proteomics. Mol Cell Proteomics 2008;7:1397-1405.
180. Maeda A, Maeda T, Golczak M, Palczewski K. Retinopathy in mice induced by disrupted all-trans-retinal clearance. J Biol Chem 2008;283:26684-26693.
181. Maeda A, Maeda T, Imanishi Y, Sun W, Jastrzebska B, Hatala DA, Winkens HJ, Hofmann KP, Janssen JJ, Baehr W, Driessen CA, Palczewski K. Retinol dehydrogenase (RDH12) protects photoreceptors from light-induced degeneration in mice. J Biol Chem 2006;281:37697-37704.
182. Maeda A, Maeda T, Imanishi Y, Kuksa V, Alekseev A, Bronson JD, Zhang HB, Zhu L, Sun WY, Saperstein DA, Rieke F, Baehr W, Palczewski K. Role of photoreceptor-specific retinol dehydrogenase in the retinoid cycle in vivo. J Biol Chem 2005;280:18822-18832.
183. Weiter JJ, Delori FC, Wing GL, Fitch KA. Retinal pigment epithelial lipofuscin and melanin and choroidal melanin in human eyes. Invest Ophthalmol Vis Sci 1986;27:145-152.
184. Fite KV, Bengston L, Donaghey B. Experimental light damage increases lipofuscin in the retinal pigment epithelium of Japanese quail (Coturnix coturnix Japonica). Exp Eye Res 1993;57:449-460.
185. Katz ML, Stone WL, Dratz EA. Fluorescent pigment accumulation in retinal pigment epithelium of antioxidant-deficient rats. Invest Ophthalmol Vis Sci 1978;17:1049-1058.
186. Feeney-Burns L, Berman ER, Rothman H. Lipofuscin of human retinal pigment epithelium. Am J Ophthalmol 1980;90:783-791.
187. Katz ML, Christianson JS, Gao CL, Handelman GJ. Iron-induced fluorescence in the retina: dependence on vitamin A. Invest Ophthalmol Vis Sci 1994;35:3613-3624.
188. Hahn P, Qian Y, Dentchev T, Chen L, Beard J, Harris ZL, Dunaief JL. Disruption of ceruloplasmin and hephaestin in mice causes retinal iron overload and retinal degeneration with features of age-related macular degeneration. Proc Natl Acad Sci U S A 2004;101:13850-13855.
189. Finnemann SC, Leung LW, Rodriguez-Boulan E. The lipofuscin component A2E selectively inhibits phagolysosomal degradation of photoreceptor phospholipid by the retinal pigment epithelium. Proc Natl Acad Sci U S A 2002;99:3842-3847.
190. Sugano E, Tomita H, Ishiguro SI, Isago H, Tamai M. Nitric oxide-induced accumulation of lipofuscin-like materials is caused by inhibition of cathepsin S. Curr Eye Res 2006;31:607-616.
191. Sundelin SP, Nilsson SEG. Lipofuscin-formation in retinal pigment epithelial cells is reduced by antioxidants. Free Radic Biol Med 2001;31:217-225.
192. Hadziahmetovic M, Dentchev T, Song Y, Haddad N, He X, Hahn P, Pratico D, Wen R, Harris ZL, Lambris JD, Beard J, Dunaief JL. Ceruloplasmin/hephaestin knockout mice model morphologic and molecular features of AMD. Invest Ophthalmol Vis Sci 2008;49:2728-2736.
193. Justilien V, Pang JJ, Renganathan K, Zhan X, Crabb JW, Kim SR, Sparrow JR, Hauswirth WW, Lewin AS. SOD2 knockdown mouse model of early AMD. Invest Ophthalmol Vis Sci 2007;48:4407-4420.
194. Katz ML, Gao CL, Rice LM. Long-term variations in cyclic light intensity and dietary vitamin a intake modulate lipofuscin content of the retinal pigment epithelium. J Neurosci Res 1999;57:106-116.
195. Katz ML, Eldred GE, Robison WG, Jr. Lipofuscin autofluorescence: evidence for vitamin A involvement in the retina. Mech Ageing Dev 1987;39:81-90.
196. Lorenz B, Wabbels B, Wegscheider E, Hamel CP, Drexler W, Preising MN. Lack of fundus autofluorescence to 488 nanometers from childhood on in patients with early-onset severe retinal dystrophy associated with mutations in RPE65. Ophthalmology 2004;111:1585-1594.
197. Katz ML, Wendt KD, Sanders DN. RPE65 gene mutation prevents development of autofluorescence in retinal pigment epithelial phagosomes. Mech Ageing Dev 2005;126:513-521.
198. Katz ML, Redmond TM. Effect of RPE65 knockout on accumulation of lipofuscin fluorophores in the retinal pigment epithelium. Invest Ophthalmol Vis Sci 2001;42:3023-3030.
199. Kim SR, Fishkin N, Kong J, Nakanishi K, Allikmets R, Sparrow JR. RPE65 Leu450Met variant is associated with reduced levels of the retinal pigment epithelium lipofuscin fluorophores A2E and iso-A2E. Proc Natl Acad Sci U S A 2004;101:11668-11672.
200. Feeney-Burns L, Hilderbrand ES, Eldridge S. Aging human RPE - morphometric analysis of macular, equatorial, and peripheral cells. Invest Ophthalmol Vis Sci 1984;25:195-200.
201. Delori FC, Goger DG, Dorey CK. Age-related accumulation and spatial distribution of lipofuscin in RPE of normal subjects. Invest Ophthalmol Vis Sci 2001;42:1855-1866.
202. Mata NL, Weng J, Travis GH. Biosynthesis of a major lipofuscin fluorophore in mice and humans with ABCR-mediated retinal and macular degeneration. Proc Natl Acad Sci U S A 2000;97:7154-7159.
203. Mata NL, Tzekov RT, Liu XR, Weng J, Birch DG, Travis GH. Delayed. dark-adaptation and lipofuscin accumulation in abcr+/- mice: Implications for involvement of ABCR in age-related macular degeneration. Invest Ophthalmol Vis Sci 2001;42:1685-1690.
204. Radu RA, Yuan Q, Hu J, Peng JH, Lloyd M, Nusinowitz S, Bok D, Travis GH. Accelerated accumulation of lipofuscin pigments in the RPE of a mouse model for ABCA4-mediated retinal dystrophies following vitamin A supplementation. Invest Ophthalmol Vis Sci 2008;49:3821-3829.
205. Allikmets R, Singh N, Sun H, Shroyer NE, Hutchinson A, Chidambaram A, Gerrard B, Baird L, Stauffer D, Peiffer A, Rattner A, Smallwood P, Li YX, Anderson KL, Lewis RA, Nathans J, Leppert M, Dean M, Lupski JR. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nature Genetics 1997;15:236-246.
206. Cremers FPM, van De Pol DJR, van Driel M, den Hollander AI, van Haren FJJ, Knoers N, Tijmes N, Bergen AAB, Rohrschneider K, Blankenagel A, Pinckers A, Deutman AF, Hoyng CB. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt's disease gene ABCR. Hum Mol Genet 1998;7:355-362.
207. Martinez-Mir A, Paloma E, Allikmets R, Ayuso C, del Rio T, Dean M, Vilageliu L, Gonzalez-Duarte R, Balcells S. Retinitis pigmentosa caused by a homozygous mutation in the Stargardt disease gene ABCR. Nature Genetics 1998;18:11-12.
208. Delaey JJ, Verougstraete C. Hyperlipofuscinosis and subretinal fibrosis in Stargardts disease. Retin-J Retin Vitr Dis 1995;15:399-406.
209. Birnbach CD, Jarvelainen M, Possin DE, Milam AH. Histopathology and immunocytochemistry of the neurosensory retina in fundus flavimaculatus. Ophthalmology 1994;101:1211-1219.
210. Kolb H, Gouras P. Electron microscopic observations of human retinitis pigmentosa, dominantly inherited. Invest Ophthalmol 1974;13:487-498.
211. Katz ML, Drea CM, Eldred GE, Hess HH, Robison WG. Influence of early photoreceptor degeneration on lipofuscin in the retinal-pigment epithelium. Exp Eye Res 1986;43:561-573.
212. Katz ML, Eldred GE. Retinal light damage reduces autofluorescent pigment deposition in the retinal pigment epithelium. Invest Ophthalmol Vis Sci 1989;30:37-43.
213. Thanos S. Sick photoreceptors attract activated microglia from the ganglion cell layer: a model to study the inflammatory cascades in rats with inherited retinal dystrophy. Brain Res 1992;588:21-28.
214. Nandrot EF, Kim Y, Brodie SE, Huang X, Sheppard D, Finnemann SC. Loss of synchronized retinal phagocytosis and age-related blindness in mice lacking alphavbeta5 integrin. J Exp Med 2004;200:1539-1545.
215. Rakoczy PE, Zhang D, Robertson T, Barnett NL, Papadimitriou J, Constable IJ, Lai CM. Progressive age-related changes similar to age-related macular degeneration in a transgenic mouse model. Am J Pathol 2002;161:1515-1524.
216. Hoppe G, Marmorstein AD, Pennock EA, Hoff HF. Oxidized low density lipoprotein-induced inhibition of processing of photoreceptor outer segments by RPE. Invest Ophthalmol Vis Sci 2001;42:2714-2720.
217. Okubo A, Sameshima M, Unoki K, Uehara F, Bird AC. Ultrastructural changes associated with accumulation of inclusion bodies in rat retinal pigment epithelium. Invest Ophthalmol Vis Sci 2000;41:4305-4312.
218. Hoppe G, O'Neil J, Hoff HF, Sears J. Accumulation of oxidized lipid-protein complexes alters phagosome maturation in retinal pigment epithelium. Cell Mol Life Sci 2004;61:1664-1674.
219. Crabb JW, O'Neil J, Miyagi M, West K, Hoff HF. Hydroxynonenal inactivates cathepsin B by forming Michael adducts with active site residues. Protein Sci 2002;11:831-840.
220. Brunk UT, Wihlmark U, Wrigstad A, Roberg K, Nilsson SE. Accumulation of lipofuscin within retinal pigment epithelial cells results in enhanced sensitivity to photooxidation. Gerontology 1995;41:201-211.
221. Bergmann M, Schutt F, Holz FG, Kopitz J. Inhibition of the ATP-driven proton pump in RPE lysosomes by the major lipofuscin fluorophore A2-E may contribute to the pathogenesis of age-related macular degeneration. Faseb J 2004;18:562-564.
222. Bermann M, Schutt F, Holz FG, Kopitz J. Does A2E, a retinoid component of lipofuscin and inhibitor of lysosomal degradative functions, directly affect the activity of lysosomal hydrolases? Exp Eye Res 2001;72:191-195.
223. Holz FG, Schutt F, Kopitz J, Eldred GE, Kruse FE, Volcker HE, Cantz M. Inhibition of lysosomal degradative functions in RPE cells by a retinoid component of lipofuscin. Invest Ophthalmol Vis Sci 1999;40:737-743.
224. Katz ML. Is lipofuscin eliminated from cells? Response. Invest Ophthalmol Vis Sci 1999;40:2464-2464.
225. Katz ML, Rice LM, Gao CL. Reversible accumulation of lipofuscin-like inclusions in the retinal pigment epithelium. Invest Ophthalmol Vis Sci 1999;40:175-181.
226. Katz ML, Shanker MJ. Development of lipofuscin-like fluorescence in the retinal-pigment epithelium in response to protease inhibitor treatment. Mech Ageing Dev 1989;49:23-40.
227. Katz ML. Incomplete proteolysis may contribute to lipofuscin accumulation in the retinal pigment epithelium. In: Porta EA (ed), Lipofuscin and Ceroid Pigments. New York: Plenum Press; 1990:109-118.
228. Ivy GO, Kanai S, Ohta M, Smith G, Sato Y, Kobayashi M, Kitani K. Lipofuscin-like substances accumulate rapidly in brain, retina and internal organs with cysteine protease inhibition. In: Porta EA (ed), Lipofuscin and Ceroid Pigments. New York: Plenum Press; 1990:31-47.
229. Katz ML, Stientjes HJ, Gao CL, Norberg M. Bright environmental light accelerates rhodopsin depletion in retinoid-deprived rats. Invest Ophthalmol Vis Sci 1993;34:2000-2008.
230. Miceli MV, Newsome DA, Tate DJ, Sarphie TG. Pathologic changes in the retinal pigment epithelium and Bruch's membrane of fat-fed atherogenic mice. Curr Eye Res 2000;20:8-16.
231. Lorenz B, Preising MN. Best's disease. Overview of pathology and its causes. Ophthalmologe 2005;102:111-115.
232. Wabbels B, Preising MN, Kretschmann U, Demmler A, Lorenz B. Genotype-phenotype correlation and longitudinal course in ten families with Best vitelliform macular dystrophy. Graefes Arch Clin Exp Ophthalmol 2006;244:1453-1466.
233. Marmorstein AD, Stanton JB, Yocom J, Bakall B, Schiavone MT, Wadelius C, Marmorstein LY, Peachey NS. A model of best vitelliform macular dystrophy in rats. Invest Ophthalmol Vis Sci 2004;45:3733-3739.
234. Bakall B, Radu RA, Stanton JB, Burke JM, McKay BS, Wadelius C, Mullins RF, Stone EM, Travis GH, Marmorstein AD. Enhanced accumulation of A2E in individuals homozygous or heterozygous for mutations in BEST1 (VMD2). Exp Eye Res 2007;85:34-43.
235. Karan G, Lillo C, Yang Z, Cameron DJ, Locke KG, Zhao Y, Thirumalaichary S, Li C, Birch DG, Vollmer-Snarr HR, Williams DS, Zhang K. Lipofuscin accumulation, abnormal electrophysiology, and photoreceptor degeneration in mutant ELOVL4 transgenic mice: A model for macular degeneration. Proc Natl Acad Sci U S A 2005;102:4164-4169.
236. Brill E, Malanson KM, Radu RA, Boukharov NV, Wang ZY, Chung HY, Lloyd MB, Bok D, Travis GH, Obin M, Lem J. A novel form of transducin-dependent retinal degeneration: Accelerated retinal degeneration in the absence of rod transducin. Invest Ophthalmol Vis Sci 2007;48:5445-5453.
237. Ambati J, Anand A, Fernandez S, Sakurai E, Lynn BC, Kuziel WA, Rollins BJ, Ambati BK. An animal model of age-related macular degeneration in senescent Ccl-2-or Ccr-2-deficient mice. Nat Med 2003;9:1390-1397.
238. Chan CC, Ross RJ, Shen D, Ding X, Majumdar Z, Bojanowski CM, Zhou M, Salem N, Jr., Bonner R, Tuo J. Ccl2/Cx3cr1-deficient mice: an animal model for age-related macular degeneration. Ophthalmic Res 2008;40:124-128.
239. Tuo J, Bojanowski CM, Zhou M, Shen D, Ross RJ, Rosenberg KI, Cameron DJ, Yin C, Kowalak JA, Zhuang Z, Zhang K, Chan CC. Murine ccl2/cx3cr1 deficiency results in retinal lesions mimicking human age-related macular degeneration. Invest Ophthalmol Vis Sci 2007;48:3827-3836.
240. Tanaka N, Ikawa M, Mata NL, Verma IM. Choroidal neovascularization in transgenic mice expressing prokineticin 1: An animal model for age-related macular degeneration. Mol Ther 2006;13:609-616.
241. Majji AB, Cao JT, Chang KY, Hayashi A, Aggarwal S, Grebe RR, de Juan E. Age-related retinal pigment epithelium and Bruch's membrane degeneration in senescence-accelerated mouse. Invest Ophthalmol Vis Sci 2000;41:3936-3942.
242. Schmitz-Valckenberg S, Holz FG, Bird AC, Spaide RF. Fundus autofluorescence imaging: review and perspectives. Retina 2008;28:385-409.
243. Delori FC, Dorey CK, Staurenghi G, Arend O, Goger DG, Weiter JJ. In vivo fluorescence of the ocular fundus exhibits retinal pigment epithelium lipofuscin characteristics. Invest Ophthalmol Vis Sci 1995;36:718-729.
244. Dorey CK, Wu G, Ebenstein D, Garsd A, Weiter JJ. Cell loss in the aging retina - relationship to lipofuscin accumulation and macular degeneration. Invest Ophthalmol Vis Sci 1989;30:1691-1699.
245. Holz FG, Bindewald-Wittich A, Fleckenstein M, Dreyhaupt J, Scholl HPN, Schmitz-Valckenberg S. Progression of geographic atrophy and impact of fundus autofluorescence patterns in age-related macular degeneration. Am J Ophthalmol 2007;143:463-472.
246. Weiter JJ, Delori F, Dorey CK. Central sparing in annular macular degeneration. Am J Ophthalmol 1988;106:286-292.
247. Rozanowska M, Jarvis-Evans J, Korytowski W, Boulton ME, Burke JM, Sarna T. Blue light-induced reactivity of retinal age pigment - in vitro generation of oxygen-reactive species. J Biol Chem 1995;270:18825-18830.
248. Boulton M, Dontsov A, Jarvis-Evans J, Ostrovsky M, Svistunenko D. Lipofuscin is a photoinducible free-radical generator. J Photochem Photobiol B-Biol 1993;19:201-204.
249. Rozanowska M, Korytowski W, Rozanowski B, Skumatz C, Boulton ME, Burke JM, Sarna T. Photoreactivity of aged human RPE melanosomes: A comparison with lipofuscin. Invest Ophthalmol Vis Sci 2002;43:2088-2096.
250. Dontsov AE, Glickman RD, Ostrovsky MA. Retinal pigment epithelium pigment granules stimulate the photo-oxidation of unsaturated fatty acids. Free Radic Biol Med 1999;26:1436-1446.
251. Rozanowska M, Pawlak A, Rozanowski B, Skumatz C, Zareba M, Boulton ME, Burke JM, Sarna T, Simon JD. Age-related changes in the photoreactivity of retinal lipofuscin granules: Role of chloroform-insoluble components. Invest Ophthalmol Vis Sci 2004;45:1052-1060.
252. Gaillard ER, Atherton SJ, Eldred G, Dillon J. Photophysical Studies on Human Retinal Lipofuscin. Photochem Photobiol 1995;61:448-453.
253. Reszka K, Eldred GE, Wang RH, Chignell C, Dillon J. The photochemistry of human retinal lipofuscin as studied by EPR. Photochem Photobiol 1995;62:1005-1008.
254. Pawlak A, Wrona M, Rozanowska M, Zareba M, Lamb LE, Roberts JE, Simon JD, Sarna T. Comparison of the aerobic photoreactivity of A2E with its precursor retinal. Photochem Photobiol 2003;77:253-258.
255. Sparrow JR, Boulton M. RPE lipofuscin and its role in retinal-pathobiology. Exp Eye Res 2005;80:595-606.
256. Lamb LE, Simon JD. A2E: A component of ocular lipofuscin. Photochem Photobiol 2004;79:127-136.
257. Roberts JE, Kukielczak BM, Hu DN, Miller DS, Bilski P, Sik RH, Motten AG, Chignell CF. The role of A2E in prevention or enhancement of light damage in human retinal pigment epithelial cells. Photochem Photobiol 2002;75:184-190.
258. Kanofsky JR, Sima PD, Richter C. Singlet-oxygen generation from A2E. Photochem Photobiol 2003;77:235-242.
259. Sparrow JR, Nakanishi K, Parish CA. The lipofuscin fluorophore A2E mediates blue light-induced damage to retinal pigmented epithelial cells. Invest Ophthalmol Vis Sci 2000;41:1981-1989.
260. Shamsi FA, Boulton M. Inhibition of RPE lysosomal and antioxidant activity by the age pigment lipofuscin. Invest Ophthalmol Vis Sci 2001;42:3041-3046.
261. Wassell J, Davies S, Bardsley W, Boulton M. The photoreactivity of the retinal age pigment lipofuscin. J Biol Chem 1999;274:23828-23832.
262. Zhou JL, Jang YP, Kim SR, Sparrow JR. Complement activation by photooxidation products of A2E, a lipofuscin constituent of the retinal pigment epithelium. Proc Natl Acad Sci U S A 2006;103:16182-16187.
263. Jang YP, Matsuda H, Itagaki Y, Nakanishi K, Sparrow JR. Characterization of peroxy-A2E and furan-A2E photooxidation products and detection in human and mouse retinal pigment epithelial cell lipofuscin. J Biol Chem 2005;280:39732-39739.
264. Sparrow JR, Vollmer-Snarr HR, Zhou JL, Jang YP, Jockusch S, Itagaki Y, Nakanishi K. A2E-epoxides damage DNA in retinal pigment epithelial cells - Vitamin E and other antioxidants inhibit A2E-epoxide formation. J Biol Chem 2003;278:18207-18213.
265. Ben-Shabat S, Itagaki Y, Jockusch S, Sparrow JR, Turro NJ, Nakanishi K. Formation of a nonaoxirane from A2E, a lipofuscin fluorophore related to macular degeneration, and evidence of singlet oxygen involvement. Angew Chem-Int Edit 2002;41:814-817.
266. Radu RA, Mata NL, Bagla A, Travis GH. Light exposure stimulates formation of A2E oxiranes in a mouse model of Stargardt's macular degeneration. Proc Natl Acad Sci U S A 2004;101:5928-5933.
267. Wang Z, Keller LMM, Dillon J, Gaillard ER. Oxidation of A2E results in the formation of highly reactive aldehydes and ketones. Photochem Photobiol 2006;82:1251-1257.
268. Gaillard ER, Avalle LB, Keller LMM, Wang Z, Reszka KJ, Dillon JP. A mechanistic study of the photooxidation of A2E, a component of human retinal lipofuscin. Exp Eye Res 2004;79:313-319.
269. Dillon J, Wang Z, Avalle LB, Gaillard ER. The photochemical oxidation of A2E results in the formation of a 5,8,5 ',8 '-bis-furanoid oxide. Exp Eye Res 2004;79:537-542.
270. Avalle LB, Wang Z, Dillon JP, Gaillard ER. Observation of A2E oxidation products in human retinal lipofuscin. Exp Eye Res 2004;78:895-898.
271. Kim SR, Jang YP, Jockusch S, Fishkin NE, Turro NJ, Sparrow JR. The all-trans-retinal dimer series of lipofuscin pigments in retinal pigment epithelial cells in a recessive Stargardt disease model. Proc Natl Acad Sci U S A 2007;104:19273-19278.
272. Rozanowska M, Bakker L, Boulton ME, Pawlak A, Rozanowski B. Protective effects of phosphatidylethanolamine (PE) against (photo)toxicity to the retinal pigment epithelium (RPE) of peroxidized docosahexaenoate (DHE). 2nd Meeting of the International Society for Ocular Cell Biology (ISOCB) 2-5 September 2008, San Diego, CA, USA; 2008.
273. Sliney DH. Exposure geometry and spectral environment determine photobiological effects on the human eye. Photochem Photobiol 2005;81:483-489.
274. Penn JS, Williams TP. Photostasis: regulation of daily photon-catch by rat retinas in response to various cyclic illuminances. Exp Eye Res 1986;43:915-928.
275. Schremser JL, Williams TP. Rod outer segment (ROS) renewal as a mechanism for adaptation to a new intensity environment. 1. Rhodopsin levels and ROS length. Exp Eye Res 1995;61:17-23.
276. Reme CE, Wolfrum U, Imsand C, Hafezi F, Williams TP. Photoreceptor autophagy: effects of light history on number and opsin content of degradative vacuoles. Invest Ophthalmol Vis Sci 1999;40:2398-2404.
277. Wiegand RD, Joel CD, Rapp LM, Nielsen JC, Maude MB, Anderson RE. Polyunsaturated fatty acids and vitamin E in rat rod outer segments during light damage. Invest Ophthalmol Vis Sci 1986;27:727-733.
278. Penn JS, Anderson RE. Effect of light history on rod outer segment membrane composition in the rat. Exp Eye Res 1987;44:767-778.
279. Penn JS, Naash MI, Anderson RE. Effect of light history on retinal antioxidants and light damage susceptibility in the rat. Exp Eye Res 1987;44:779-788.
280. Penn JS, Anderson RE. Effects of light history on the rat retina. Progr Retinal Res 1991;11:75-98.
281. Handelman GJ, Dratz EA. The role of antioxidants in the retina and retinal pigment epithelium and the nature of prooxidant-induced damage. Adv Free Radical Biol Med 1986;2:1-89.
282. Beatty S, Koh HH, Henson D, Boulton M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv Ophthalmol 2000;45:115-134.
283. Wrona M, Rozanowska M, Sarna T. Zeaxanthin in combination with ascorbic acid or alpha- tocopherol protects ARPE-19 cells against photosensitized peroxidation of lipids. Free Radic Biol Med 2004;36:1094-1101.
284. Wrona M, Korytowski W, Rozanowska M, Sarna T, Truscott TG. Cooperation of antioxidants in protection against photosensitized oxidation. Free Radic Biol Med 2003;35:1319-1329.
285. Ellis EM. Reactive carbonyls and oxidative stress: potential for therapeutic intervention. Pharmacol Ther 2007;115:13-24.
286. Maeda A, Crabb JW, Palczewski K. Microsomal glutathione S-transferase 1 in the retinal pigment epithelium: Protection against oxidative stress and a potential role in aging. Biochemistry 2005;44:480-489.
287. Hayes JD, Flanagan JU, Jowsey IR. Glutathione transferases. Annu Rev Pharmacol Toxicol 2005;45:51-88.
288. Naash MI, Nielsen JC, Anderson RE. Regional distribution of glutathione peroxidase and Glutathione-S-transferase in adult and premature human retinas. Invest Ophthalmol Vis Sci 1988;29:149-152.
289. Sarna T, Rozanowska M. Phototoxicity to the eye. In: Jori G, Pottier RH, Rodgers MAJ, Truscott TG (eds), Photobiology in Medicine. New York: Plenum Press; 1994:125-142.
290. van Norren D, Schellekens P. Blue light hazard in rat. Vision Res 1990;30:1517-1520.
291. Heck M, Schadel SA, Maretzki D, Bartl FJ, Ritter E, Palczewski K, Hofmann KP. Signaling states of rhodopsin - Formation of the storage form, metarhodopsin III, from active metarhodopsin II. J Biol Chem 2003;278:3162-3169.
01/28/09
03/09/09