BACTERIAL BIOLUMINESCENCE
Biochemistry and Molecular Biology
Leo Yen-Cheng Lin1 and Edward A. Meighen2
1Progen Biotech Inc.
126-11782 River Road
Richmond, BC V6X1Z7 Canada
leoyenchenglin@hotmail.com
2Department of Biochemistry, McGill University
3655 Promenade Sir William Osler, Montreal, QC, H3G1Y6, Canada
janmeighen@yahoo.com
Luminous Bacteria - Habitation and Taxonomy
Luminous bacteria are the most widely distributed light-emitting organisms with the majority existing in seawater and the remainder living in the terrestrial or freshwater environment. While most species of luminescent bacteria are capable of living free, the majority are found in nature associated in symbiosis with host organisms (Figures 1 to 4) (i.e., fishes, squids, crabs, nematodes, etc...).

Figure 1. Pinecone fish utilize luminous bacteria, colonized in the ventral cavity, to illuminate the surroundings as well as for intra-species communication.
In symbiosis, the bacteria are nourished with readily available food sources for growth, and at the same time the host utilizes the adopted illumination to communicate, to attract prey, and to masquerade itself from predators.

Figure 2. The deep sea Angler fish carries luminous bacteria in a light emitting rod, which attracts prey to the front of its mouth.
However, there are certain species of luminescent bacteria, which are obligatory symbionts, requiring unique nutritional supplements, which are exclusively available from the host. Although the presence of these obligatory symbionts has been detected, they are not separable from their host, and therefore are unable to be cultured in the laboratory for further study.
There are three major genera, into which most luminous bacteria are classified; Photobacterium, Vibrio, and Photorhabdus. Species existing in the marine environment are mainly categorized into the Photobacterium and Vibrio genera, and the terrestrial species are classified into the Photorhabdus (previously designated as Xenorhabdus) genus. Species within the Photobacterium genus are generally light organ symbionts of marine animals, whereas the Vibrio species exist as free-living forms as well as symbionts in the sea.
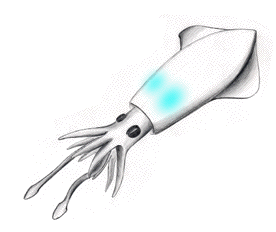
Figure 3. Luminous bacteria reside in symbiosis on a pair of light organs in the mantle body of the squid. Utilization of the illumination function is believed to frighten nearby predators, allowing the squid to escape.
Many luminous bacteria are parasitic, with Photobacterium and Vibrio families infecting marine crustacea, and Photorhabdus luminescens infecting terrestrial insects, such as caterpillars, with nematodes as the intermediate host for the bacteria (Figure 4). In addition, free-living luminous bacteria that are dispersed in the seawater can often be found in both the gut tract and on the skin surface of almost all marine animals as non-specific parasites.
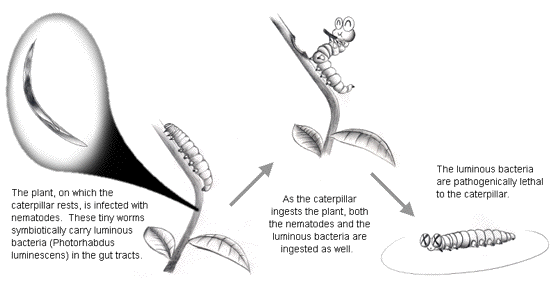
Figure 4. Infection with the terrestrial luminous bacteria (Photohabdus luminescens) is pathogenically lethal to the caterpillar.
For luminous bacteria residing within the gut tracts of marine animals, the extra-cellular chitinase produced on the cell wall of all luminous bacteria facilitate the decomposition of the ingested chitin (Figure 5) (e.g., from the exoskeletons of crustacea).
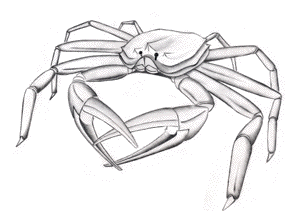
Figure 5. Luminous bacteria are non-specific parasites of crustacea. The exoskeletons of Tanner crabs are the sites of colonization of luminous bacteria, whose infection causes lesions on the surface of its appendages.
Each species of luminous bacteria differs in a number of properties, including the specific growing conditions (nutritional requirements and growth temperature), and the reaction kinetics of the luciferase involved in light generation; however, all luminous bacteria are rod-shaped, gram-negative microorganisms with flagella facilitating motion. Luminous bacteria are also facultative anaerobes capable of growth when the supply of molecular oxygen is limited. Despite the physiological diversity among different species of luminous bacteria, all luminescent microorganisms utilize highly homologous biochemical machineries to produce light. The onset and the energy output of this light-producing molecular machinery are tightly regulated under a central signaling pathway. The text as well as the Figures below will describe the essential biochemical components of bacterial bioluminescence, and the sequential molecular interactions/reactions along the reaction pathway. The final sections will walk briefly through the signaling pathways, by which the induction and the repression of the illumination function in luminescent bacteria are carefully controlled by cellular factors and intercellular communication.
Biochemistry of the Bacterial Bioluminescence Reaction - Bacterial Luciferase and the Light Color
Bacterial luciferase is the enzyme that catalyzes light emission at the heart of bacterial bioluminescence. However, the catalytic machinery involved in continuous light production in luminous bacteria includes not only bacterial luciferase, but also the enzymes that supply and regenerate the substrates of bacterial luciferase.
The DNA sequences coding the proteins in the luminescent system are termed the lux genes. Bacterial luciferase is a heterodimer, composed of two different polypeptides, designated alpha and beta (of molecular mass 40 kDa and 37 kDa, respectively, and encoded by the luxA and luxB genes, respectively. The active site is located within the subunit
(Figure 6).
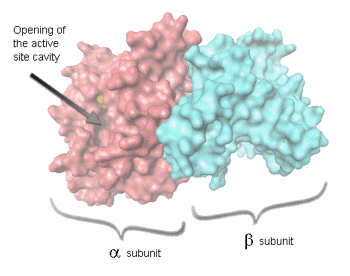
Figure 6. Bacterial luciferase structure.
In the absence of the beta subunit, the alpha subunit alone functions inefficiently with a poor light yield. As the crystal structure of V. harveyi luciferase reveals extensive interactions and complex binding patterns between several side chains and backbone amides of the alpha and beta subunits (Figure 7), the function of the beta subunit has been assumed to act as a supporting scaffold assisting in the conformational change of the subunit during the catalysis.
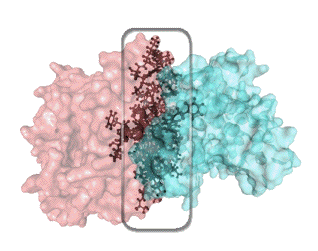
Figure 7. The network of extensive inter-subunit interactions, highlighted in the rectangular box, and involving ionic attractions, hydrogen bonds, and hydrophobic contacts, is responsible for the assembly of the functional bacterial luciferase.
The substrates of bacterial luciferase are reduced flavin mononucleotide (FMNH2), molecular oxygen, and long chain fatty aldehyde (Figure 8). The excess energy, which is liberated from the oxidation of FMNH2 and aldehyde concomitant with the reduction of molecular oxygen, is released as blue/green light emission (
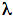 MAX ~ 490 nm) (Figure 8). Luciferases function well with long chain aldehydes of eight carbons or longer, with tetradecanal presumed to be the natural aldehyde used in vivo in the luminescent bacteria.
MAX ~ 490 nm) (Figure 8). Luciferases function well with long chain aldehydes of eight carbons or longer, with tetradecanal presumed to be the natural aldehyde used in vivo in the luminescent bacteria.
Figure 8. The net chemical equation of the bacterial luciferase catalyzed reaction.
The characteristic color indicates the energy level of the photon that was produced when the excited electron on the flavin chromophore returns to the ground state. Researchers in the field have discovered that flavin analogs with substituted atoms in the chromophore moiety resulted in different luciferase emission colors.
In addition, it has also been shown that point mutations at the flavin chromophore's binding site distorts the color emission spectrum of bacterial bioluminescence, indicating that the distinctive emission color depends not only on the chromophore that emits the photon, but also the electronic nature of the chromophore-binding microenvironment in luciferase. Aside from bacterial luciferase, some luminous bacteria carry fluorescent proteins to modulate the emission color, distinguishing themselves from other strains. Interaction of blue fluorescent protein (also known as the lumazine protein) in Photobacterium phosphoreum and Photobacterium leiognathi with the respective luciferases yields photon emission at higher energy (
MAX ~478 nm) corresponding to a blue color. Similarly, the presence of the yellow fluorescence protein in the strain Y-1 of V. fischeri results in yellow light emission (MAX ~545 nm). The participation of the fluorescence proteins in the lucifrease-catalyzed reaction also alters the reaction kinetics of bacterial luciferase. The structural/molecular basis for the shift/change in emission colors favors the ability of fluorescence protein to interact (e.g., stabilize or destabilize) with the high-energy luciferase reaction intermediate (e.g., excited chromophore) resulting in different colors of light emission. Nevertheless, the energy input is clearly provided by the oxidation of FMNH2, and aldehyde and bacterial luciferase. In order for light emission to occur for prolonged period of times, substrates must be supplied continuously to bacterial luciferase. As the time frame and the level of product generation in enzymatic reactions are limited by the availability of the substrates, the constant light emission in luminous bacteria must therefore be maintained by several different enzymes continuously generating the substrates for the bioluminescence reaction. Those enzymes that replenish the aldehyde substrate are coded on the lux operon; in particular, the fatty acid reductase, a multienzyme complex, whose lux genes (luxC, luxD, and luxE) immediately flank the luxA and luxB genes of luciferase (Figure 9).
Other genes including luxF, luxG, and luxH, whose functions are neither clearly defined nor apparently necessary for bioluminescence are also found in some lux operons.
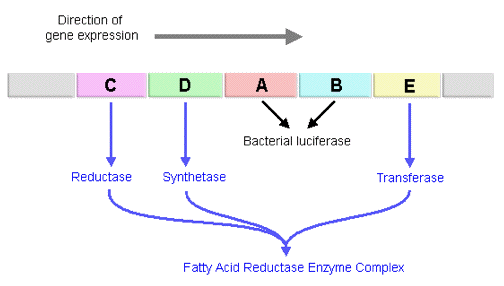
Figure 9. The arrangement of the luxCDABE open reading frames.
Fatty Aldehyde Synthesis and Recycling in Luminous Bacteria - Fatty Acid Reductase
Although a structural model of the fatty acid reductase at atomic resolution to visualize how fatty acid is exchanged among subunits of this multienzyme complex and modified toward the fatty aldehyde in three-dimensional space is not available, separation and isolation of fatty acid reductase into individual subunits has allowed the identification of the function of each component in the protein complex. Fatty acid reductase is a large protein with approximately 500 kilodaltons of molecular mass. This large complex is comprised of 12 polypeptides (four copies of polypeptides expressed by each of the luxC, luxD, and luxE genes) (Figure 11). Although the designated letters for this set of lux genes go from C to E, the progression of aldehyde synthesis does not follow the same direction (Figure 9). LuxD encodes the transferase, which transfers the acyl moiety of fatty acyl-ACP, acyl-CoA, and other activated acyl donors derived from the universal fatty acid biosynthetic pathway, to the hydroxyl group of a serine on the transferase, followed by the conversion of the ester to a fatty acid through hydrolysis (Figure 10). The transferase has a high specificity for acyl derivatives with chain lengths of fourteen carbons, consistent with tetradecanal being the aldehyde used in the bioluminescent reaction in vivo.
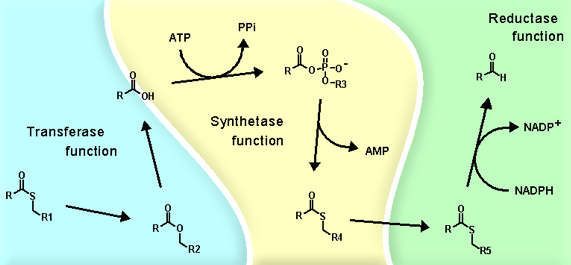
Figure 10. The channeling of substrates among the transferase (blue), the synthetase (yellow), and the reductase (green) in the fatty acid reductase enzyme complex.
The fatty acid produced by the transferase is then channeled to the synthetase (LuxE), which activates the carboxylate function with ATP to form a mixed anhydride of acyl-AMP (Figure 10). The acyl group on the anhydride intermediate, acyl-AMP, then reacts with the reactive thiol of cysteine in the synthetase, forming a covalent linkage between the acyl group and the synthetase and release of AMP (Figure 10).
Through a reciprocal trans-esterification reaction between a thiol group on the synthetase and a thiol group on the reductase (LuxC), the fatty acyl group exchanges the covalent bonding partner from synthetase to reductase (Figure 10). The reductase then breaks the thiol-ester linkage by reduction with the hydride of NADPH to release fatty aldehyde, and return itself to its original free enzyme state (Figure 10). The subunits of fatty acid reductase are organized in a radial fashion with a central core of four reductase subunits, which are weakly complexed with each individual synthetase subunit. The transferases (LuxD) branch from the synthetase at the outermost periphery in the fatty acid reductase complex, and dissociate very readily (Figure 11).
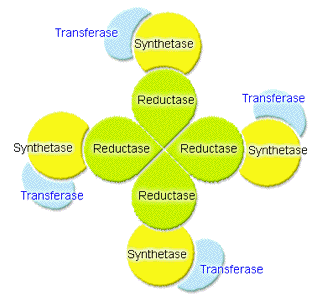
Figure 11. The association map of the transferase, synthetase, and reductase units in the fatty acid reductase enzyme complex.
The advantage of such complex formation is the efficient channeling of the reaction intermediate from within one subunit to the other without the exposure of the hydrophobic hydrocarbons of the reaction intermediates into the aqueous cytosolic environment of the luminous bacteria (Figure 11). In addition, the arrangement of transferase, synthetase, and reductase in a radial fashion channels the reaction material through different microenvironments that stabilize the acyl intermediate at various stages of reaction as the material is processed from the aqueous periphery, where the acyl group is received, towards the center of the complex where the reductase reduces the fatty acyl group to aldehyde (Figure 10). As all proteins in the fatty acid reductase complex have a high specificity for tetradecanoyl groups, the final product is tetradecanal.
Recharging the FMNH2 - FMN Reductase
Flavin mononucleotide (FMN) is an electron carrier present in the electron transport chains of both eukaryotic and prokaryotic cells. FMN is capable of donating and receiving one electron through radical formation, or two electrons with hydride transfer (Figure 12).
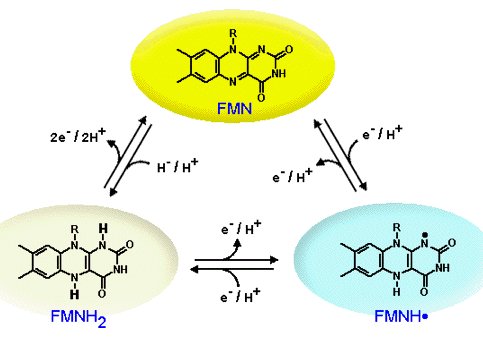
Figure 12. The 3 oxidation states of flavin mononucleotide: FMN, the oxidized state, is highlighted within the yellow oval. FMNH., the half reduced or the radical state, is highlighted in the blue oval. FMNH2, the fully reduced or the hydroquinone state, is highlighted in the pale oval.
FMN is derived from riboflavin (also known as Vitamin B2), and both riboflavin and FMN are common nutrients essential for the survival of both eukaryotic and prokaryotic cells (Figure 13).
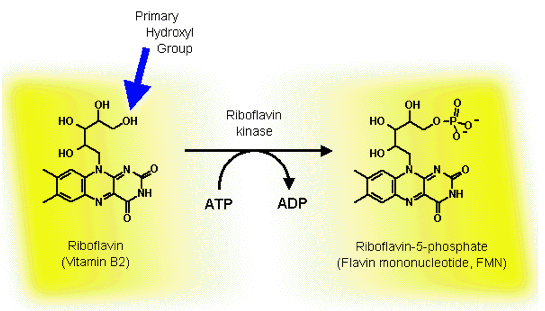
Figure 13. The transfer of the phosphate group from ATP to riboflavin. The blue arrow highlights the primary hydroxyl group, to which riboflavin kinase adds the phosphate, and distinguishes it from the other secondary hydroxyl groups on the ribityl side chain.
The fact that the conversion of a nonluminous bacterium, such as Escherichia coli, to a light-emitter requires only the insertion of the luxCDABE genes, encoding the bacterial luciferase and the fatty acid reductase complex, into the cell, indicates that FMNH2 is readily provided from the electron transport chain in all bacteria. Although the biosynthesis of riboflavin and FMN in luminous bacteria is carried out in multiple steps by enzymes that are not encoded by the lux gene system, these enzymes are generally present in bacteria, as riboflavin and FMN synthesis is required for bacterial growth. The reduction of FMN to FMNH2 is accomplished by the addition of the hydride from NAD(P)H in a reaction catalyzed by a FMN/NAD(P)H oxidoreductase (also known as FMN reductase) in luminous bacteria (Figure 14).
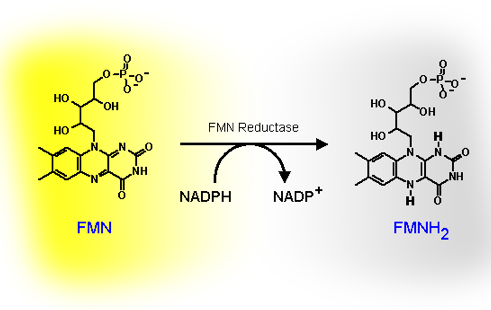
Figure 14. The reduction of FMN to FMNH2, coupled with the oxidation of NAD(P)H to NAD(P)+, is catalyzed by FMN reductase.
The crystal structures of dimeric FMN reductase from both V. harveyi and V. fischeri reveal that FMN reductase is a homodimer containing two FMNs bound at the symmetrical inter-subunit interface (Figure 15).
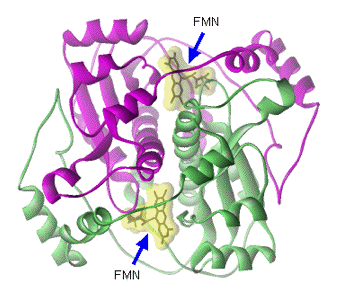
Figure 15. Two FMNs, which are highlighted in yellow clouds, are embraced at the inter-subunit interface between the two identical subunits that are colored in red and green.
Recently, researchers in the field have carried out extensive kinetic investigations in an attempt to elucidate how FMNH2 is channeled from the FMN reductase to the bacterial luciferase. Based on the strong binding affinities of FMN reductase toward both FMN and FMNH2, and the tightly anchored FMN binding geometries in the crystal structure of FMN reductase, investigators have speculated that the transfer of FMNH2 from FMN reductase to bacterial luciferase occurs by transient formation of a luciferase-FMN reductase complex in luminous bacteria, rather than through free diffusion of FMNH2 dispatched into the cytosol (Figure 16 and 17). This would result in a very efficient recycling and reduction of FMN to FMNH2 for use by bacterial luciferase (Figure 17).
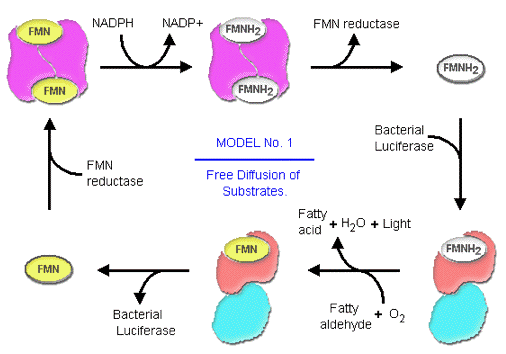
Figure 16. The uptake of substrate from the cytosolic environment is one of the two possible modes, by which bacterial luciferases have access to FMNH2.
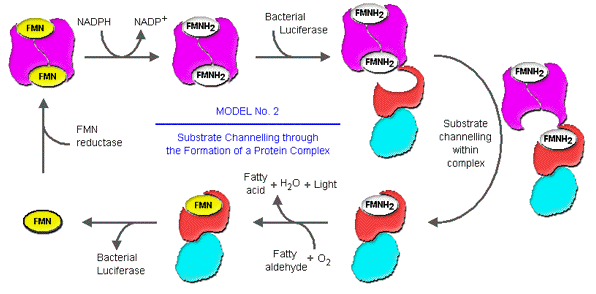
Figure 17. The direct channeling of the FMNH2 from FMN reductase to the bacterial luciferase within the transient reductase-luciferase complex is the second possible mode of substrate delivery to the luciferase.
The Universal Oxidant in Bioluminescence - Molecular Oxygen
A critical component in the biochemistry of bacterial bioluminescence is molecular oxygen, which is supplied from the external cellular environment. Without the input of molecular oxygen, luminous bacteria cannot emit light. In the bacterial luciferase-catalyzed reaction, the energy expenditure on the reduction of molecular oxygen to a peroxy reaction intermediate, and then ultimately to water serves as a trigger for releasing the potential energy from the oxidation of both FMNH2 and fatty aldehyde in the form of photon emission (Figure 18).
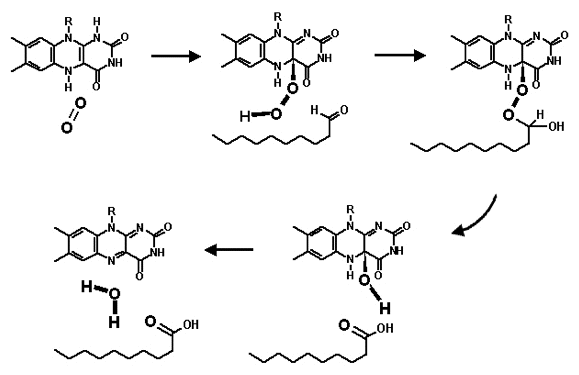
Figure 18. The chemical mechanism of the substrate-substrate interactions in the luciferase catalyzed reaction.
In addition to bacterial bioluminescence, all the other biological luminescence systems (such as fireflies, coelenterates and dinoflagellates) utilize molecular oxygen as the obligatory oxidant in their luminescence biochemistry, and the processes involved in the reduction of the molecular oxygen serves as an energy sink, draining the reducing power of the substrates. This leads to the formation of unstable high energy intermediates, which dissipate the potential energy of the excited chromophore in the form of light. In this regard, molecular oxygen can be considered to serve as a key to unleash the energy deposited in FMNH2 and fatty aldehyde for bacterial bioluminescence.
The Utilization of Bacterial Bioluminescence as a Biosensor and Reporter of Gene Expression.
As the primary function of bacterial luciferase is to catalyze the emission of light, this feature together with generation of the aldehyde substrate by fatty acid reductase can be successfully produced in other bacteria, by the transfer of the luxCDABE genes, which convert nonluminescent bacteria into light emitters (Figure 19).
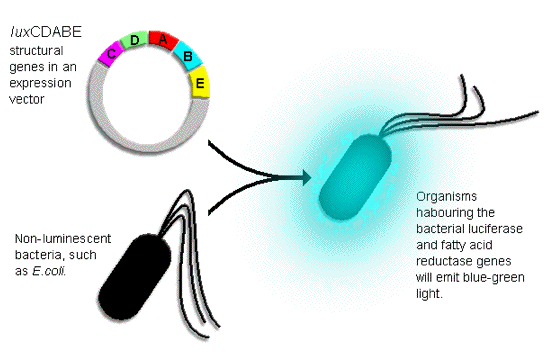
Figure 19. The "insertion" of the foreign luxCDABE structural genes into the organism allows the fatty acid reductase enzyme complex and luciferase to be expressed, and the function of luciferase confers the organism the ability to emit light.
The adaptation of light as an accessory feature of normally nonluminescent bacteria has provided researchers an easy alternative to measure and detect the growth and living conditions of bacteria (Figure 19). The utilization of bacterial bioluminescence is also one of several methods used in the detection of pathogenic bacteria in human food sources. By culturing a food sample in the presence of a recombinant bacteriophage carrying the luxCDABE insert, one can readily determine the presence of bacterial contamination in the food source. In addition, the light emitting property of the luxCDABE genes has been employed as a reporter of gene expression for studying regulatory controls involved in affecting the efficiency of RNA polymerase in initiation and transcription at different promoters. Then the luxCDABE genes are under the control of an environmentally regulated promoter (e.g., promoters whose efficiency is highly sensitive to the level of mercury, arsenic, or other pollutants), the structural lux genes can function as a biosensor, whose expression will monitor the presence of toxic waste in the environment. In the pharmaceutical industry, genetically modified bacteria carrying the lux luminous system have been utilized to evaluate the efficacy of antibiotics in fighting against bacterial infections in mammals; with animals such as mice, pigs, and monkeys serving as potential human models. In this screening procedure, the intensity of luminescence in the infected organs/tissues will decrease as the bacteria are killed by the antibiotics; therefore, bacterial bioluminescence serves as an indicator of bacterial growth allowing the proper dosages of antibiotics to be determined and effective treatment to be established.
Regulation of the luxCDABE Expression in Luminous Bacteria - Quorum Sensing
The workload of the machinery that emits light in luminous bacteria is under stringent regulatory controls. In order for the bright emission to occur, not only a high level of the expression of the luxCDABE genes is required to form bacterial luciferase and fatty acid reductase, but also the synthesis of the substrates of bacterial luciferase is essential to maintain the light emission over long time periods. Hence, the emission of light by luminous bacteria is highly dependent on the growth and the environment. In the laboratory set-up, luminous bacteria growing in liquid media at low cell density give off a minimal amount of light, because of the dormancy of the expression of luxCDABE genes and the deficiency in the level of substrate for the bacterial luciferase reaction (Figure 20). From the mid to the end of the exponential growth period, the intensity of the light emission rises dramatically as the result of the rapid accumulation of the synthesized substrates and enzymes from the activation of the expression of the luxCDABE genes (Figure 20).
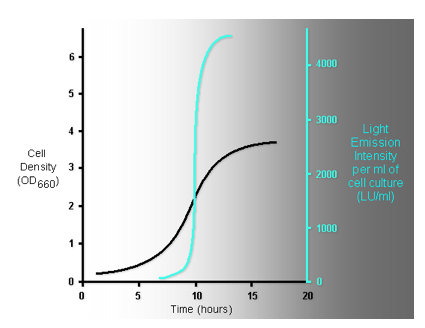
Figure 20. The superimposition of the growth curve of luminous bacteria (colored in black), and the glowing intensity from the cell culture measured along the growth (colored in light blue) indicates that the mid-log phase of the growth of luminous bacteria is the "switch point" where "the light is turned on".
The increase in the expression of the luxCDABE genes occurs through a mechanism analogous to an autocatalytic chemical reaction, in which the products of the reaction also catalyze the reaction from which they were derived, leading to the generation of more products and therefore more catalysts (Figure 21).
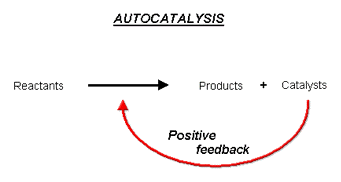
Figure 21. Autocatalysis is the catalysis of the reaction by one of the reaction product(s), which exponentially and automatically increases the reaction rate as the reaction proceeds.
In bacterial bioluminescence the catalysts that induce the expression of the luxCDABE gene are regulatory proteins, and a small chemical compound, which is commonly called the autoinducer (Figure 22).
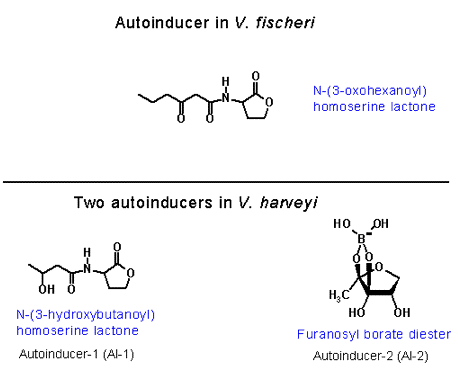
Figure 22. Chemical structures of the autoinducers of luminous bacteria.
The autoinducer is a small metabolic product, which freely diffuses across the cellular membrane, and is excreted to the extracellular environment during the early stage of cell growth (Figure 23).
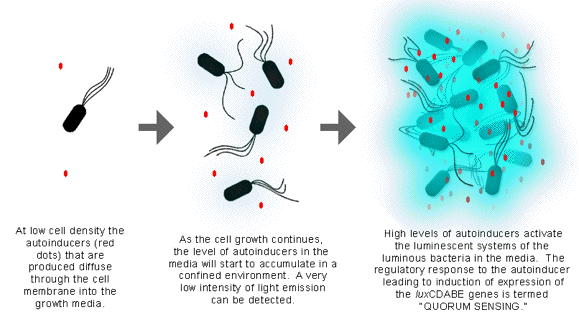
Figure 23. The confined environment, in which the produced autoinducers can accumulate, is the key to the activation of the quorum sensing mechanism.
It is known that marine luminescent bacteria that live free in the ocean do not emit light, while bacteria living as symbionts in marine organisms and/or in a localized, confined environment emit a high level of light. Therefore, in order for the light emission to occur, luminous bacteria have to grow in a confined, nutrition-rich environment, in which the autoinducer can be accumulated. When the concentration of the autoinducer increases to a certain level in the extracellular environment, the autoinducers will activate the luminous system of luminous bacteria. This process has been called "quorum sensing", as the bacteria literally sense when a sufficient concentration of bacteria has been reached based on the accumulation of autoinducer (Figure 23).
Most investigations have focused on the quorum sensing systems of Vibrio fischeri and Vibrio harveyi, as these luminescent bacteria have been extensively investigated. Although both V. fischeri and V. harveyi utilize the luxCDABE structural genes (coding for bacterial luciferase and fatty acid reductase) to generate light, the mechanism of the activation of luxCDABE gene expression is somewhat different between the two species.
The Autocatalytic Mechanism Involved in the Regulation of V. fischeri luxCDABE Expression.
The autoinducer that turns on the bioluminescent system of V. fischeri is 3-oxohexanoyl-homoserine lactone, synthesized from S-adenosylmethionine and 3-oxohexanoyl-ACP (acyl carrier protein) by the autoinducer synthetase coded by the luxI gene (Figure 24).
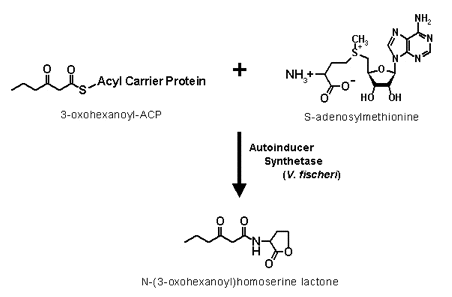
Figure 24. The amide linkage formation from 3-oxohexanoyl and methionine groups, and the lactonization of the methionine side chain are the two reaction steps involved in the formation of the homoserine lactone autoinducers.
When a critical concentration of 3-oxohexanoyl-homoserine lactone is established in the extracellular environment, autoinducer starts to accumulate inside the luminous bacteria and interacts with the regulatory protein coded by luxR (Figure 24).
One notable feature about the bioluminescence system in V. fischeri is the organization of the regulatory lux genes in controlling the induction of bacterial bioluminescence, particularly the bi-directional RNA polymerase promoter binding site between the oppositely oriented luxR operon and the contiguously linked, polycistronic operon containing the luxICDABE genes (Figure 25).
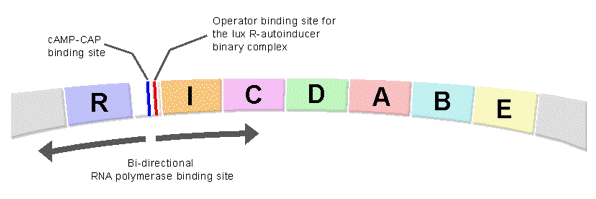
Figure 25. The arrangement of the regulatory elements in the upstream region of the lux structural genes in V. fischeri.
Binding of the activation complex (autoinducer-LuxR) to the operator site in the regulatory region between the luxR and luxICDABE operons along with a cAMP-CRP (cAMP-cAMP receptor protein) complex at a CRP binding site, leads to RNA polymerase being recruited to the bi-directional promoter site to transcribe the downstream lux genes (Figure 25). As a consequence of the organization of these lux genes in the genome, the activation complex formed between the autoinducer and the regulatory protein (LuxR) accelerates not only the expression of luxCDABE but also the expression of more LuxR regulatory protein and LuxI autoinducer synthetase. Within a short time interval the feedback amplification on the subsequent rounds of lux gene expression have produced a large number of luciferase and fatty acid reductase molecules as well as increased levels of autoinducer and regulatory protein (LuxR), resulting in an exponential increase in the emission intensity per cell and the bright emission of the luminous bacteria observed after autoinduction (Figure 21, 23, and 27). The utilization of an autocatalytic mechanism in accelerating gene expression is not exclusive to luminous bacteria, as investigators in other fields have found that many non-luminous bacteria use similar quorum-sensing mechanisms to orchestrate the onset and enhancement of gene expression.
While the activation and the luxCDABE gene expression in V. fischeri have been well studied, the mechanism by which the luminescence appears to be repressed at very high cell densities is not well understood. This decrease in light intensity may reflect a limitation in substrate availability; alternatively, the autoinducer-LuxR activation complex may bind to a low-affinity operator-like binding site causing repression.
Comparison of the Regulation Mechanisms of V. fischeri and V. harveyi
In contrast to the well-studied regulatory pathway controlling V. fischeri luminescence, the control mechanisms involved in regulating the onset of luminescence in other species of luminous bacteria have not been fully explored. The regulation of V. harveyi luminescence has been the recent target of interest. Although autoinducers and the luxCDABE genes (luciferase and fatty acid reductase) are the common structural elements essential for the onset of light emission in most luminous bacteria, the control mechanism by which the level of the expression of luxCDABE is regulated in V. harveyi is extremely complex compared to that of V. fischeri. In contrast to the luxI/R pair of regulatory genes in directing luxCDABE expression in V. fischeri, a multitude of at least eight regulatory lux genes are involved in signal transduction in controlling the onset of V. harveyi luminescence (Figure 27). However, the signal transduction in V. harveyi from the autoinducer quorum sensor in the extracellular environment to the operon in the cell is functionally homologous to the luxI/R of V. fischeri in luminescence activation. The purpose of the partitioning of the integrated function over various regulatory components may be due to the coupling of the luminescence regulation to one or more metabolic pathways and fine-tuning the level of luminescence emission in V. harveyi in response to nutritional signals. It therefore is necessary to elucidate the function of each regulatory element involved in the signal relay system to determine the potential interactions of these regulatory components with other cellular metabolic pathways and their contributions to the modulation of the level of luminescence emission. Consequently, the partitioned functions of the regulatory components in V. harveyi in relation to the integrated luxI/R in V. fischeri will be briefly described here. In contrast to the closely linked and contiguous luxR and luxICDABE operons in the V. fischeri genome, all the regulatory lux genes involved in controlling the luxCDABE expression are scattered over the V. harveyi genome. In addition, there are two signaling transduction pathways, responding to two different autoinducers in V. harveyi luminescence regulation. The autoinducer, N-(3-hydroxybutanoyl)homoserine lactone (Figure 22), is produced by the autoinducer synthetase encoded by luxM and the other autoinducer, a furanosyl borate diester (Figure 26), is synthesized by the luxS autoinducer synthetase.
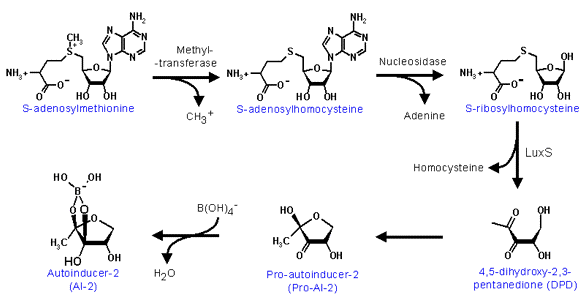
Figure 26. The reaction mechanisms involved in the biosynthesis of furanosyl borate diester, the autoinducer-2 in V. harveyi.
Although the luxI synthetase of V. fischeri synthesizes a homoserine lactone derivative that is similar to that produced by the LuxM synthetase of V. harveyi, neither LuxM nor LuxS share sequence homology to autoinducer synthetase (LuxI) of V. fischeri. In contrast to the formation of the transcriptional activator complex (LuxI autoinducer and LuxR regulatory protein), which directly acts on the luxCDABE operon to activate transcription in V. fischeri, the autoinducers excreted into the extracellular environment by V. harveyi bind to corresponding autoinducer receptors (LuxM autoinducer binds the extracellular domain of the LuxN transmembrane protein; LuxS autoinducer binds the extracellular LuxP subunit of the LuxP/Q transmembrane protein complex) on membrane proteins, which have autoinducer receptor domains in the extracellular environment and kinase/phosphatase domains in the intracellular environment (Figure 27). The binding of autoinducers to the corresponding receptor domains then causes a conformational change in the intracellular domains, activating the intracellular phosphatase activity of LuxN and LuxP/Q transmembrane signal mediators (Figure 27). From this point on, the signal converges, and both the phosphatase domains of LuxN and LuxP/Q dephosphorylate the LuxU regulatory protein, which then dephosphorylates LuxO, removing the repression from the luxCDABE genes. LuxO is a sigma-54 regulator and is believed to stimulate synthesis of a repressor of the lux operon and/or luxR. Once the LuxO regulatory protein is dephosphorylated, the repression on luxCDABE expression is relieved, leaving LuxR as the activator of the luxCDABE operon (Figure 27). However, it is important to emphasize that V. harveyi LuxR does not possess any sequence or structural homology to V. fischeri LuxR nor does the V. harveyi LuxR directly interact with the autoinducer. It is interesting to notice that V. harveyi utilizes an extensive signal transduction mechanism involving multiple steps of phosphorylation and dephosphorylation to deliver the membrane-diffusible autoinducer signal from the extracellular environment to the LuxR activator in the cell, while the LuxR protein of V. fischeri directly binds the autoinducer to activate luxCDABE expression (Figure 27).
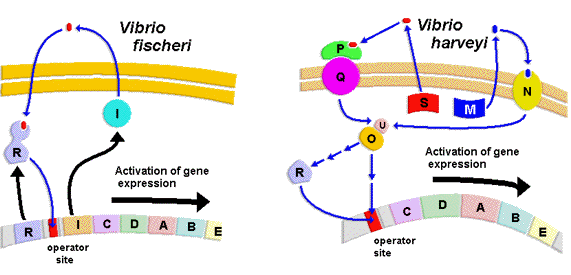
Figure 27. The comparison of the different mechanisms involved in the regulation of the expression of the lux structural genes in V. fischeri and V. harveyi.
As both the phosphorylation and dephosphorylation are integral activities of various signal transductions pathways involved in controlling cellular growth, the transfer of the quorum-sensing signal by a phospho-transduction system to the lux operon provides an excellent opportunity to integrate metabolic energy with the quorum sensing regulation in luminous bacteria.
Conclusion
The phenomena of bioluminescence in nature have always been a focus of human attention. Prior to the identification of luminous organisms, the presence of even a dim luminous glow in the dark frightened countless numbers of individuals, who perceived the glimmer as ghosts or supernatural spirits. However, the curiosity of scientists to solve the mysteries of nature led to the identification of many luminous organisms responsible for the light emission in different environmental settings. The isolation of luminous bacteria from marine and terrestrial environments, and the characterization of their properties have resulted in the identification of a number of luminous bacterial species classified into several genera. All luminescent bacteria utilize FMNH2, O2, and long fatty aldehyde as substrates for the bioluminescence reaction catalyzed by luciferase (LuxAB), with the fatty acid reductase complex (LuxCDE) synthesizing the long chain aldehyde substrate of tetradecanal. A NAD(P)H-dependent FMN reductase found in most if not all bacteria generates the FMNH2 substrate.
Consequently, only the five genes in the lux operon, luxCDABE, are needed to produce light emission, even in bacteria that normally do not emit light, and thus provide the opportunity to utilize the bacterial lux system as a light emitting sensor in many bacteria.
The regulation of the induction of expression of the luxCDABE genes in luminescent bacteria at high cell density has led to the development of molecular prototypes for quorum sensing, an important new regulatory mechanism signaling bacterial crowding, and now found controlling key secondary metabolic pathways in many nonluminescent bacteria. Advances in understanding the molecular mechanisms, structures and regulation of the lux enzymes and genes of luminescent bacteria has answered many of the questions, but why bacteria utilize the energy of the cell to emit light in this exergonic process, and how the luminescent system interfaces at the molecular level with the metabolic and energy supplying pathways in the bacteria still remains a mystery.
Interactive Exercise
Bacterial luciferase is one of the few enzymes that has been extensively investigated over the past few decades. The reason behind its continuously-received attention in science, particularly in enzyme research, is the complexity of the substrate-substrate and substrate-enzyme interaction along the reaction pathway.
A crucial factor, which effectively assists researchers in resolving and understanding the complex intermolecular interaction in the luciferase catalyzed reaction, is the unique architecture of the active site in the 3-dimensional structure of bacterial luciferase. However, before taking the advantage of this structural information in functional inferences, scientists not only have to accurately locate the ligand-binding site, but also have to be able to visualize the conformation of the protein bound ligand in coordination with all the active site structural features in the surrounding environment. Because the crystal structure of bacterial luciferase does not contain bound substrates, researchers in this field have recently built the flavin into the active site of bacterial luciferase with molecular modeling.
The structural information, derived from the molecular modeling investigation not only correlates with several experimental results already existing in the literature but also allows researchers to expand their research projects to new areas. In this exercise you will walk through the experiments, and analyze all the results supplied to you to derive a hypothesis, which was actually used in the development of the recently proposed flavin-luciferase complex model.
Suggestion: The following step-by-step procedure outlines the highlights of events, which actually took place during the development of the proposed flavin-luciferase complex model. You should read the graphics and results, and process the information before proceeding to the next step. The purpose of this walkthrough is to put you in simulation with the events, which occurred during the actual research project.
Step 01: Inspecting the active site of bacterial luciferase. The active site space of bacterial luciferase is represented by the blue-bubble at the center of Figure 28. The outline of the blue-blob is the contour surface of the active site cavity, created by amino acid residues.
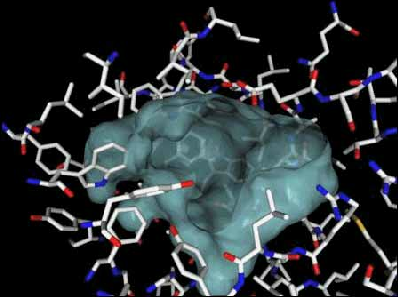
Figure 28. The active site of bacterial luciferase, represented as a blue blob, is a large open cavity. The opening at the top of blue blob is the entrance through which the substrates (FMNH2, fatty long chain aldehyde, and molecular oxygen) have access through. In order for the enzyme catalysis to occur, the enzyme-substrate binding interaction has to be established first. The formation of enzyme-substrate binary complex is primarily determined by the structural complementarity (e.g., shape, size, and partial charge properties) between the binding partners.
As you can see from the above figure, the volume and the shape of the active site cavity (blue bubble) is very large and deep. The purpose for this large empty space is obvious, because bacterial luciferase has to have a "roomy pocket" to accommodate two large substrates (the reduced flavin mononucleotide and the fatty long chain aldehyde) and one oxygen molecule. Secondly, it can also be seen that there is no protruding structural elements dividing this cavity into designated substrate binding site for each of the substrates, foreshadowing the complexity involved in the intermolecular interactions between substrates in coordination with the active site pocket.
Step 02: Relating the inorganic phosphate ion to the phosphate group of flavin mononucleotide. The presence of an inorganic phosphate ion in the active site provides scientists a good guess/hint about the position (Figure 29), to which the phosphate group of flavin is most likely to anchor in the active site.
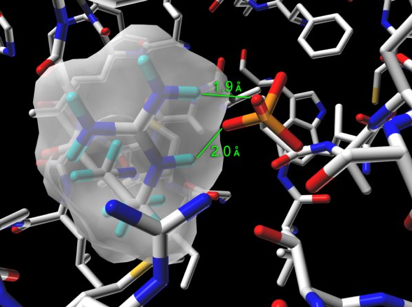
Figure 29. The bound inorganic phosphate ion in the active site of bacterial luciferase (Protein Data Bank entry: 1BRL.pdb) establishes salt bridge binding interaction with the guanidinium group of Arg-107. The guanidinium group of Arg-107 is highlighted with its opaque solvent accessible surface in the figure.
However, it is important for an investigator (you, as a scientist) to confirm this hypothesis before assigning the geometrical coordinates of the phosphate group of flavin to the location where the inorganic phosphate ion resides in the crystal structure (Figure 30).
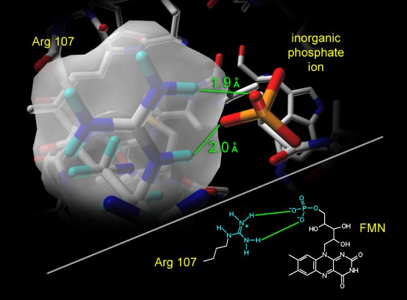
Figure 30. Could the location of this inorganic phosphate ion be the same site to which the phosphate group of flavin (e.g., substrate FMNH2 or product FMN) anchors? Experiments are needed to test and validate this hypothesis.
To support this hypothesis, site directed mutagenesis and substrate analogs were used in combination with kinetic analysis to establish the direct interaction between the negatively charged phosphate group of flavin and the positively charged guanidinium group of Arg107 (Figure 31). The strategies include the mutation of the Arg107, whose guaninidium group is positively charged (pKa ~ 12), to a glutamate, whose carboyxlate side chain is negatively chaged (pKa ~4). The use of riboflavin, a FMN analog that lacks the negatively charged phosphate group, in combination with the changes in the side chain electronic property at position 107, were used to assess the validity of the hypothesis.
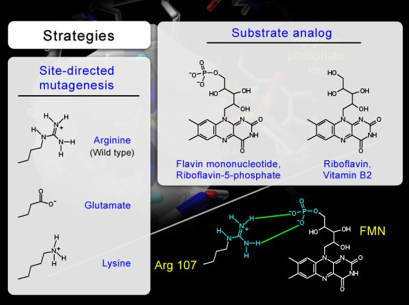
Figure 31. To support the hypothesis and confirm the proposed intermolecular interaction between the guanidinium group of Arg107 and the phosphate group of flavin, experiments are designed with two different strategies. The objective is to use site-directed mutagenesis and substrate analog to verify the complementarity between the phosphate group of flavin and the side chain at position 107 in bacterial luciferase.
The activity of the luciferases, measured against both FMNH2 and riboflavin shows that the glutamate mutant, R107E, prefers riboflavin to FMNH2, indicating that the negatively charged carboxylate side chain of R107E is repelling the negatively charged phosphate group of FMNH2 (Figure 32). The reversal in the substrate preference in R107E mutant demonstrates the existence of interaction between the side chain at position 107 and the phosphate group of FMNH2.
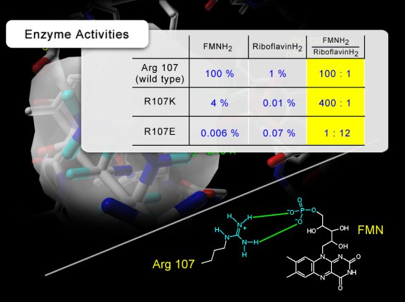
Figure 32. The activities of luciferase mutants measured in the presence of FMNH2 and riboflavinH2. In the case of R107E mutant, the specificity of luciferase for the flavin substrate (FMNH2/riboflavinH2) is reversed. In addition, the data shows that the mutation at position 107 significantly affects the substrate specificity of luciferase for FMNH2 and riboflavinH2, which clearly supports the direct interaction established between the phosphate group of FMN and the guanidinium group of Arg 107.
In addition, the insensitivity of the glutamate mutant, R107E, to the increasing inorganic phosphate ion concentration indicates that the negatively charged carboxylate side chain of R107E effectively repels the inorganic phosphate ion from entering the phosphate-binding site (Figure 33). As these results establish an excellent correlation between the effect of mutation and the site of mutation, the function of the side chain at position 107 in binding the phosphate group of flavin substrate was established, and the hypothesis was also supported.
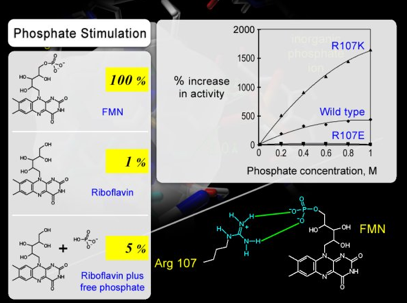
Figure 33. RiboflavinH2 is a poor substrate because of the absence of phosphate group. The activity of riboflavin can be stimulated by 5-folds by simply adding inorganic phosphate ion in the in vitro bioluminescence assay. However, luciferase with R107E mutation is insensitive to inorganic phosphate ion, indicating that the glutamate substitution at position 107 (R107E mutation) repels the inorganic phosphate ion in solution. This result again supports the hypothesis of Arg107 being the phosphate anchor for flavin substrate.
Step 03: Searching for the most probable flavin binding geometry.
Figure 34 shows the overlapping of all the binding conformations of flavin found during the molecular modeling. Here, the "messiness" of the superimposed flavin models represents that the active site cavity has been thoroughly searched for the flavins with geometrically possible conformations. However, as several literature references, published over the past few decades, have clearly indicated that the enzyme bound flavin is tightly coordinated with little vibration in motion in the ligand binding site, the correct binding geometry must be one of the models shown above. Therefore, a screening procedure for selecting the correct model must be devised.
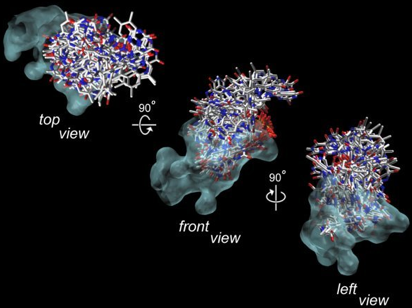
Figure 34. The opaque blue-blob is the surface contour of the active site in luciferase. Within the active site (blue-blob), all the possible binding conformations of flavin are superimposed. This slide simply shows you that the active site cavity is spacious, and the computer simulation has searched through all the possible binding geometries in the active site cavity (blue-blob). The correct binding geometry of flavin is one of the conformations shown above, but which one? Without further information to support the selection procedure for identifying the correct model, it is more difficult than "finding a needle in a haystack."
Step 04: Identifying the pharmacophoric information from the structure-activity data of flavin analogs. Figure 35 shows the results extracted from a published article. As the linker group, which connects the phosphate to the catalytic isoalloxazine, changes in length, the activity also changes. The dramatic change from full enzyme activity with phospho-butyl-flavin to hundred-fold lower activity with phospho-propyl-flavin indicates that flavin-enzyme interaction is highly dependent on the length of the linker moiety, which is highlighted in blue in the above Figure. That is, with the phosphate group of flavin anchored at the phosphate binding site in the enzyme, and following the projection of the catalytic isoalloxazine group into the active site cavity, the linker group must have sufficient length for the catalytic isoalloxazine group to adapt an active binding geometry in the active site. The result in Figure 36 shows that the minimal linker size required for this binding process to successfully occur is 4-carbons long. The dramatic difference in activity between the linker size of 3-carbons unit and 4-carbons unit implies that the length of the linker is the constraining structural element controlling the overall function of the molecule.
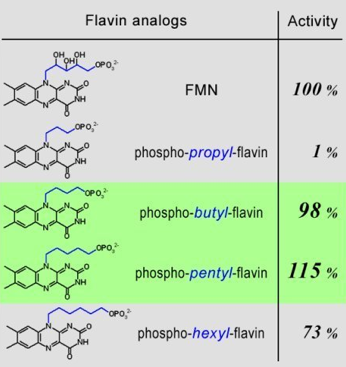
Figure 35. Pharmacophore - the important structural features that determine the molecular functions. For luciferase the length of linker between flavin and phosphate is a pharmacophore, and the optimal size is 4 or 5 carbon units long. Can this information be used to identify the correct conformation of flavin predicted in the computer-assisted molecular dynamic simulations?
Step 05: Developing a selection procedure. In the schematic graph (Figure 36), the yellow cylinder represents the phosphate-binding site. The pie-shaped sector, extending from the phosphate-binding site, represents the field, to which the catalytic isoalloxazine group of flavin is allowed to reach with the given number of carbons in the linker moiety. The pharmacophoric information, extracted from the actual biochemical data, indicates that the active binding geometry occurs when the flavin has a minimum of 4-carbons in the linker moiety. With all the information in mind, the active binding geometry of the catalytic isoalloxazine moiety must be common and easily accessible by the fully active flavin analogs (i.e., with a 4-carbon and 5-carbon unit linker). In addition, this specific binding geometry of the catalytic isoalloxazine must be absent in the pool of models built with phospho-propyl-flavin. Therefore, multiple cycles of conformational searches for flavin analogs with 3, 4, and 5 carbons unit linkers were carried out. By selecting the isoalloxazine conformations, which are common among phospho-butyl-flavin and phospho-pentyl-flavin and FMN, followed by eliminating the conformers that are within the search field of phospho-propyl-flavin, researchers effectively narrowed down the number of possible models to one.
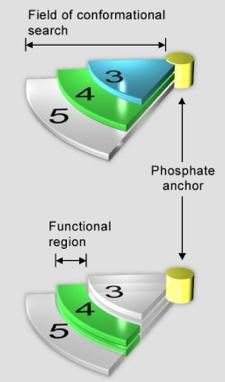
Figure 36. The selection procedure for identifying the correct conformation of flavin from the models generated in molecular modeling. The yellow cylinder represents the origin, and serves as an anchoring point for the phosphate group of flavin. The pie-shaped extensions (labeled 3, 4, and 5) represent the conformational space, to which the flavin can be modeled, in the active site of luciferase. Essentially, the longer the linker group in flavin, the further the flavin can be extended away from the origin (e.g., phosphate anchor). Since the optimal length (i.e., pharmacophore) for the linker group is 4 and 5, the correct binding geometry is a conformation accessible by phospho-butyl-flavin and phosphor-pentyl-flavin. Therefore, by performing the same molecular modeling procedure for phosphor-propyl-flavin, phosphor-butyl-flavin, and phosphor-pentyl-flavin, the common binding geometries of flavin between the models of phosphor-butyl-flavin and phosphor-pentyl-flavin will be the answer.
Figure 37 shows three flavin models, which are clustered together in space. The phospho-butyl-flavin is colored in burgundy, the phospho-pentyl-flavin in colored in green, and the natural flavin, FMN, is colored in yellow. Figure 38 shows that the isoalloxazine moieties (the tricyclic moiety) of butyl-flavin, pentyl-flavin, and FMN are all in the same orientation and space. The common geometry of the isoalloxazine chromophore shown in Figure 38 is also the model that was proposed.
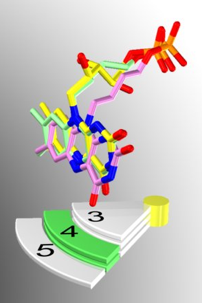
Figure 37. Artistic representation of the results produced from the molecular modeling simulations and the developed selection procedure. The yellow flavin represents the native substrate of luciferase - FMNH2. The purple flavin is phosphor-butyl-flavin, and the green flavin is phopho-pentyl-flavin. All three flavin substrates (FMNH2, phosphor-butyl-flavin, and phospho-pentyl-flavin) elicit full enzyme activities of bacterial luciferase in the in vitro bioluminescence assay. The figure shows the superimposition of the flavin moieties of FMNH2, phosphor-butyl-flavin, and phospho-pentyl-flavin. The superimposed flavin moieties were purposely placed above the green-highlighted label (4) to demonstrate the success of this project.
[NOTE: For detailed information on topics provided in this exercise, please refer to Lin et al. (2001); Modeling of the bacterial luciferase-flavin mononucleotide complex combining flexible docking with structure-activity data. Protein Sci. 10: 1563-1571.]
Suggested Reading
Baldwin, T.O., Christopher, J.A., Raushel, F.M., Sinclair, J.F., Ziegler, M.M., Fisher, A.J., and Rayment, I. (1995) Structure of bacterial luciferase. Curr. Opin. Struct. Biol. 5: 798-809.
Bassler, B.L. (2002) Small talk. Cell-to-cell communication in bacteria. Proc. Natl. Acad. Sci. USA. 99: 3129-3134.
Chen, X., Schauder, S., Potier, N., Van Dorsselaer, A., Pelczer, I., Bassler, B.L., and Hughson, F.M. (2002) Structural identification of a bacterial quorum-sensing signal containing boron. Nature 415: 545-549.
Fisher, A.J., Thompson, T.B., Thoden, J.B., Baldwin, T.O., and Rayment, I. (1996) 1.5 Å resolution crystal structure of bacterial luciferase in low salt conditions. J. Biol. Chem. 271: 21956-21968.
Hastings, J.W., and Greenberg, E.P. (1999) Quorum sensing: the explanation of a curious phenomenon reveals a common characteristic of bacteria. J. Bacteriol. 181: 2667-2669.
Koike, H., Sasaki, H., Kobori, T., Zenno, S., Saigo, K., Murphy, M.E., Adman, E.T., and Tanokura, M. (1998) 1.8 Å crystal structure of the major NAD(P)H:FMN oxidoreductase of a bioluminescent bacterium, Vibrio fischeri: overall structure, cofactor and substrate-analog binding, and comparison with related flavoproteins. J. Mol. Biol. 280: 259-273.
Mager, H.I., and Tu, S.C. (1995) Chemical aspects of bioluminescence. Photochem. Photobiol. 62: 607-614.
Meighen, E.A. (1992) Bioluminescence, bacterial. Encyclopedia of Microbiology Vol. 1: 309-319.
Meighen, E.A. (1993) Bacterial bioluminescence: organization, regulation, and application of the lux genes. FASEB J. 7: 1016-1022.
Miller, M.B., and Bassler, B.L. (2001) Quorum sensing in bacteria. Annu Rev Microbiol. 55: 165-199.
Nealson, K.H., and Hastings, J.W. (1979) Bacterial bioluminescence: its control and ecological significance. Microbiol Rev. 43: 496-518.
Tang, C.K., Jeffers, C.E., Nichols, J.C., and Tu, S.C. (2001) Flavin specificity and subunit interaction of Vibrio fischeri general NAD(P)H-flavin oxidoreductase FRG/FRase I. Arch. Biochem. Biophys. 392: 110-116.
Tanner, J.J., Lei, B., Tu, S.C., and Krause, K.L. (1996) Flavin reductase P: structure of a dimeric enzyme that reduces flavin. Biochemistry 35: 13531-13539.
Tu, S.C., and Mager, H.I. (1995) Biochemistry of bacterial bioluminescence. Photochem. Photobiol. 62: 615-624.
Wilson, T., and Hastings, J.W. (1998) Bioluminescence. Annu. Rev. Cell. Dev. Biol. 14: 197-230.
01/25/09