DETERMINING THE MECHANISM
FOR PHOTOSENSITIZED OXIDATIONS
Jeffrey R. Kanofsky
261 E Adelia Street
Elmhurst, IL 60126
kanofsky@sbcglobal.net
Introduction
Photosensitizers absorb light, generating electronically excited states.

As shown by Equations 3, 4 and 5, respectively, an excited photosensitizer may loose its excitation energy via fluorescence, via phosphorescence or via non- radiative transitions that ultimately produce heat.
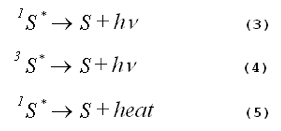
When energy loss occurs by any of these mechanisms, no photochemistry is initiated.
Photochemistry occurs when an excited photosensitizer interacts with other molecules. Most commonly, these molecules are oxygen, a solvent molecule or a target molecule added to the solvent. The excited photosensitizer may transfer either energy or charge during interactions with other molecules. The various possibilities are shown in Figure 1.
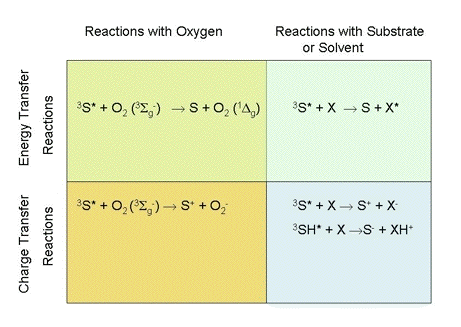
Figure 1. Mechanisms of photosensitized reactions. Here 3S* is a photosensitizer in an excited triplet state, 3SH* is another representation of the same photosensitizer, but with a potentially transferable proton indicated, X is a target molecule, and XH+ is a substrate or solvent molecule that has accepted a proton.
The mechanisms responsible for photosensitized oxidations have been classified into two categories, called Type I and Type II photochemistry. There is general agreement that reactions generating free-radicals from a target molecule or the solvent should be classified as Type I photochemistry. Similarly, there is agreement that singlet-oxygen generation should be classified as Type II photochemistry.
Authors differ on how the other mechanisms shown in Figure 1 should be classified. Some authors classify photochemical mechanisms based on the initial molecule with which the excited photosensitizer interacts (1). Photosensitizer interactions with either solvent or target molecules are called Type I photochemistry. Photosensitizer interactions with oxygen are called Type II photochemistry. Consequently, these authors would classify the formation of an electronically excited target molecule as Type I photochemistry, and superoxide generation as Type II photochemistry. Other authors feel that the distinction between Type I and Type II photochemistry should be based on whether charge transfer or energy transfer occurs (2). All charge-transfer reactions are classified as Type I photochemistry, while all energy-transfer reactions are classified as Type II photochemistry. Consequently, these authors would classify the superoxide production as Type I chemistry, and the formation of an electronically excited target molecule as Type II chemistry.
There are relatively few molecules with electronically excited states that are sufficiently low-lying so that these molecules can accept energy from commonly- used photosensitizers. Thus, the formation of an electronically-excited target or solvent molecule is an uncommon means of initiating photochemistry, particularly in biological systems. For some molecules with low-lying electronic states, such as carotenoids, the energy transferred to the molecule is often ultimately dissipated as heat rather than used to initiate a chemical reaction.
Throughout this chapter, the term singlet-oxygen will refer to the 1
 g state. There
exists a more energetic singlet state of oxygen, the 1
g state. There
exists a more energetic singlet state of oxygen, the 1 g+ state.
However, the 1g+
state rapidly relaxes to the 1g state. The lifetime of the 1g+ state is so short that
it does not appear to initiate any photochemistry (3).
g+ state.
However, the 1g+
state rapidly relaxes to the 1g state. The lifetime of the 1g+ state is so short that
it does not appear to initiate any photochemistry (3).
Considerable effort has been devoted to the development of methods that can identify the various mechanisms of photosensitized oxidations. There are many assays for singlet-oxygen generation with various degrees of sensitivity and specificity. Likewise, there are multiple tests that can detect the formation of free radicals, including superoxide. Several excellent reviews of this topic have been published previously (4-10). The major emphasis in this chapter will be on assays for singlet oxygen.
Assays for Singlet Oxygen
Assays for singlet oxygen include the measurement of singlet-oxygen
phosphorescence at 1270 nm, the detection of specific singlet-oxygen products
using various chemical traps, the inhibition of product formation using singlet-
oxygen quenchers, and the use of the deuterium-isotope solvent effect.
Singlet-oxygen phosphorescence.
Singlet oxygen is an electronically excited molecule and consequently can emit
light. The measurement of singlet-oxygen phosphorescence at 1270 nm is one of the
most specific methods for detecting singlet oxygen. The high specificity results
from the empiric observation that other emission sources with peaks close to
1270 nm are uncommon. This is probably due to the fact that very few molecules
have low-lying electronically excited states with transition energies similar to
oxygen. Since the spin restriction rule of quantum mechanics does not allow the
transition from singlet oxygen (1g) to ground-state oxygen, the intensity of the singlet-
oxygen phosphorescence is generally very weak. The low intensity of the
emission limits the sensitivity of the method. Studies of singlet-oxygen
phosphorescence can be carried out using either time-resolved or steady-state
methods.
In time resolved-studies, the photosensitizer is usually excited with a pulsed
laser. A block diagram of a typical apparatus is shown in Figure 2. The
phosphorescence is measured at right angles to the laser beam, using filters that
transmit 1270-nm radiation, but prevent scattered laser light and fluorescence
from reaching the near-infrared detector. The most commonly used near-infrared
detectors are germanium photodiodes, indium gallium arsenide photodiodes and
photomultipliers with indium phosphide/indium gallium arsenide phosphide
photocathodes. All of these detectors are sensitive to the 1270-nm radiation.
One advantage of the time-resolved method over the steady-state method is that the lifetime of the singlet-oxygen emission is measured in the time-resolved method. The measured emission lifetime can be compared with the theoretical lifetime of singlet oxygen under the conditions being studied. Agreement between the measured lifetime and the theoretical lifetime provides additional confirmation that the emission actually comes from singlet oxygen.
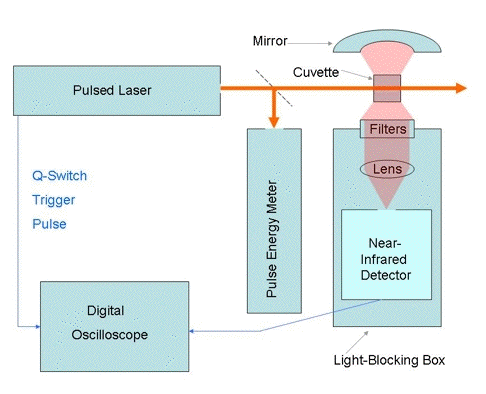
Figure 2. Apparatus for time-resolved measurements of singlet-oxygen phosphorescence.
In a homogeneous photochemical system, the formation and decay of singlet oxygen are described by Equations 6 and 7.
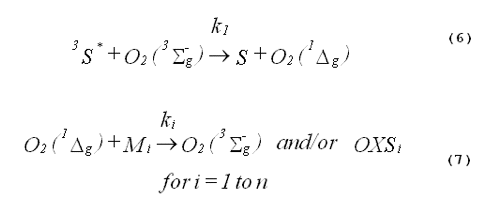

The kinetics of the excited triplet photosensitizer will be given by
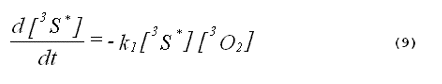
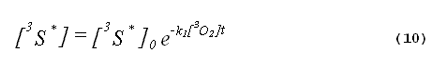
The intensity of the singlet-oxygen phosphorescence is proportional to the concentration of singlet oxygen in the system. Thus, the solution to Equation 8, multiplied by a constant, will describe the kinetics of the singlet-oxygen emission. The emission kinetics are given by Equation 11,

 is the emission lifetime, ß is the
emission rise time, and t is time (11). Under conditions where the k1[3O2] is
greater than k2, the emission rise time, ß, will be determined by the rate of
reaction of the excited photosensitizer with oxygen, and the emission lifetime, ,
will be determined by the lifetime of singlet oxygen. Under conditions where
k1[3O2] is less than k2, the emission lifetime will be determined by the rate of
reaction of the excited photosensitizer with oxygen, while the emission rise time
will be determined by the lifetime of singlet oxygen (11).
is the emission lifetime, ß is the
emission rise time, and t is time (11). Under conditions where the k1[3O2] is
greater than k2, the emission rise time, ß, will be determined by the rate of
reaction of the excited photosensitizer with oxygen, and the emission lifetime, ,
will be determined by the lifetime of singlet oxygen. Under conditions where
k1[3O2] is less than k2, the emission lifetime will be determined by the rate of
reaction of the excited photosensitizer with oxygen, while the emission rise time
will be determined by the lifetime of singlet oxygen (11).
The yield of singlet oxygen from a photosensitized reaction is proportional to the time-integrated singlet-oxygen phosphorescence intensity divided by the singlet-oxygen lifetime,
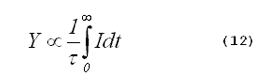
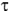 is the lifetime of singlet oxygen in the
system, and t is time. Combining Equations 11 and 12 gives (11):
is the lifetime of singlet oxygen in the
system, and t is time. Combining Equations 11 and 12 gives (11):
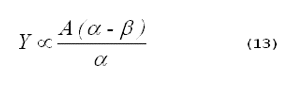 > ß).
> ß).
It is important to remember that Equation 13 cannot be used to compare the singlet-oxygen yields in different solvents. At an identical concentration of singlet oxygen, the intensity of the phosphorescence in various solvents will differ, because the radiative lifetimes of singlet oxygen vary in different solvents (12). Thus, the constant, A, will differ in different solvents.
The fluorescence of some photosensitizers extends out into the near-infrared to at least 1270 nm. An advantage of the time-resolved method over the steady-state method is that the fluorescence can sometimes be cleanly separated from the singlet-oxygen phosphorescence. In time-resolved studies, this fluorescence appears as a sharp peak coincident with the laser pulse. The apparent lifetime of the fluorescence peak is usually determined by the response time of the near-infrared detector. Large fluorescence peaks can overload near-infrared detectors or their associated electronics. This instrumental artifact limits the energy of the laser pulse that can be used, and consequently limits the sensitivity of the time-resolved method for photosensitizers with a large fluorescence at 1270 nm. Simulated data for a typical time-resolved experiment are shown in Figure 3. Note how the singlet-oxygen emission is nicely separated from the fluorescence.
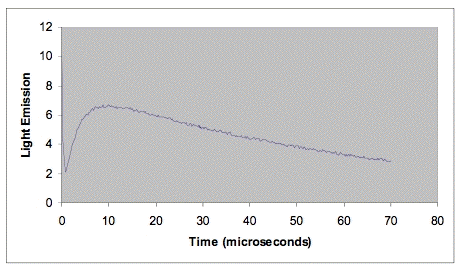
Figure 3. Data set simulating time-resolved emission from a photosensitizer in deuterium oxide. The rapidly decreasing emission at the left of the figure is due to fluorescence. The rate of increase in emission is determined by the rate at which the excited triplet photosensitizer reacts with oxygen. The decay rate is determined by the lifetime of singlet oxygen in deuterium oxide, about 68 µs.
Steady-state measurements of singlet-oxygen phosphorescence have also proved to be useful for determining mechanisms of photooxidation. The spectral resolution may be achieved with a series of interference filters, a conventional monochromator or an interferometer-based monochromator (13). ). A typical apparatus for the measurement of steady-state singlet oxygen phosphorescence is shown in Figure 4.

Figure 4. Apparatus for the measurement of steady-state singlet-oxygen phosphorescence.
Photosensitizer fluorescence in the near-infrared can potentially obscure singlet- oxygen phosphorescence in steady-state measurements. In the interpretation of emission spectra from steady-state experiments, investigators generally take advantage of the fact that photosensitizer fluorescence tends to decrease monotonically with increasing wavelength, and generally does not have any structure in the spectral region near 1270 nm. In contrast, singlet-oxygen phosphorescence shows a clear emission peak near 1270 nm. Thus, the fluorescence signal usually appears as a sloping baseline under the singlet- oxygen emission peak (13).
Singlet-oxygen traps. Singlet-oxygen traps are compounds that react with singlet oxygen to produce specific products, not generated by any other oxidants. Many traps for singlet oxygen have been developed. They differ in sensitivity, selectivity and solubility.
Cholesterol is one of the most specific traps for singlet oxygen. The reaction of singlet oxygen with cholesterol produces three specific products, 5
-cholesterol
hydroperoxide, 6-cholesterol hydroperoxide and 6ß-cholesterol hydroperoxide
(14-16). Generally, the 5-cholesterol hydroperoxide is the most abundant
product (14). In contrast, radical oxidation of cholesterol produces 7-cholesterol
hydroperoxide and 7ß-cholesterol hydroperoxide (14-17). However, the significance of two
7-cholesterol hydroperoxides is ambiguous, since 5
-cholesterol hydroperoxide, one of the oxidation products generated by singlet
oxygen, can undergo allylic rearrangement to form 7-cholesterol hydroperoxide
(14-17). Subsequently, 7-cholesterol hydroperoxide can epimerize to form 7ß-cholesterol hydroperoxide (see Figure 5)
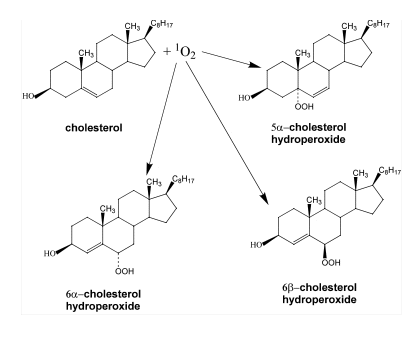
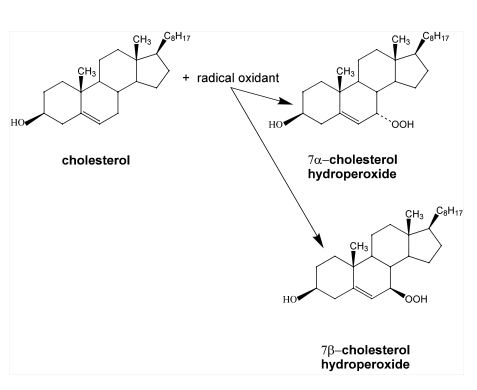
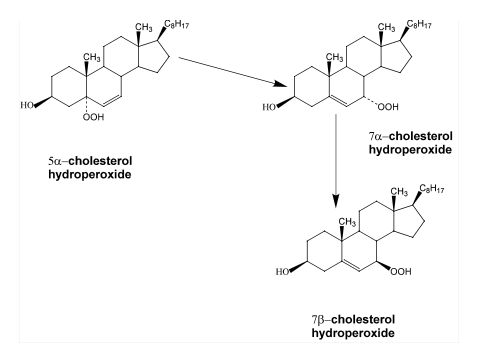
the allylic rearrangement and epimerization of
5
-cholesterol hydroperoxide.
The various cholesterol hydroperoxides can be separated with either thin-layer chromatography or HPLC. Sensitive detection methods for the cholesterol hydroperoxides have been developed that use radiolabeling of the cholesterol, chemiluminescence or electrochemical reduction with a mercury drop electrode (15, 18-19).
Cholesterol trapping has two principal limitations. First, the sensitivity of cholesterol trapping is limited due to its relatively slow rate of reaction with singlet oxygen (20). Second, cholesterol is not soluble in highly polar solvents, such as water. The solubility limitation can be overcome by preparing small beads with a thin coating of cholesterol, and placing the beads in the polar solvent during the photosensitized oxidation (21).
The reaction of singlet oxygen with various anthracene derivatives also provides a relatively specific trapping system. As shown in Figure 6, the specific product formed is an endoperoxide. Singlet oxygen reacts more rapidly with anthracenes than cholesterol, making the anthracenes more sensitive traps than cholesterol. Derivatives of anthracene have been prepared that are soluble in a wide variety of solvents including water. Thin deposits of anthracenes on the surface of small beads, or on surface of flat sheets, have also successfully been used as singlet- oxygen detectors in biological studies (22-23). One caution in the use of anthracenes is that anthracenes are photosensitizers. Thus, anthracenes cannot be used in photochemical studies at wavelengths where the anthracenes absorb light.
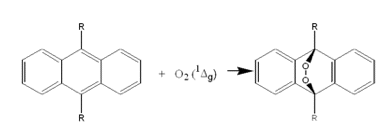
Singlet-oxygen quenchers. A singlet-oxygen quencher is a molecule that rapidly interacts with singlet oxygen either physically, producing ground-state oxygen and heat, or chemically, producing oxidation products.
Examples of molecules that are commonly used as quenchers include azide anion, 1,4-diazabicyclo[2.2.2]octane (DABCO), histidine and various carotenoids, such as ß-carotene. The principal limitation of quenchers is a lack of specificity. Most quenchers are easily oxidized substances and thus will react with many types of oxidants.
The specificity of a singlet-oxygen quencher can be improved by comparing the decrease in oxidation products caused by the addition of the quencher with the theoretical reduction expected. A formula to calculate the theoretical reduction is easily derived. Consider a photochemical system undergoing constant irradiation. The system is composed of solvent, photosensitizer, target molecules for singlet oxygen and multiple other components. Assume that the concentrations of all system components remain constant. The following two equations will apply:
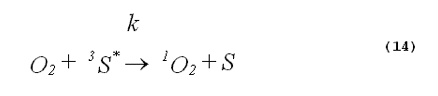
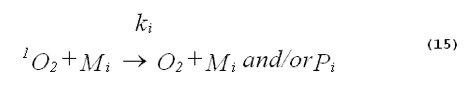
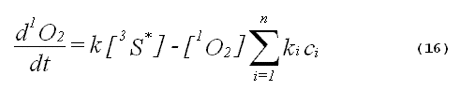
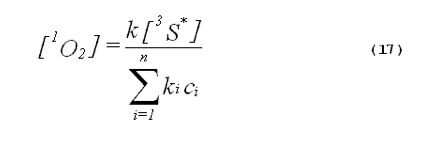
The denominator of Equation 17 can be rewritten to provide better clarity. Quenching due to the solvent is generally expressed in terms of the singlet- oxygen lifetime in the neat solvent rather than as the product of the concentration of solvent molecules and the singlet-oxygen quenching constant for the solvent. Also, we will separate the quenching component due to the added quencher from the rest of the sum. This gives,

Here
0 is the lifetime of singlet oxygen in the neat solvent, kq is the total singlet-oxygen quenching constant for the added quencher, and cq is the concentration of
the added quencher.
The rate of product formation should be directly proportional to the steady-state concentration of singlet oxygen. From Equation 18, it follows that the ratio of product formed in the absence of the added quencher (P0) to the product formed in the presence of the added quencher (P) is given by,
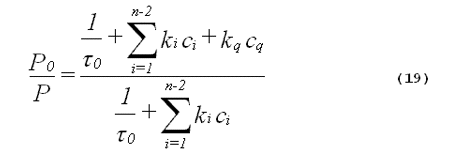
From the inspection of Equation 19, it follows that plots of P0/P against the concentration of added quencher, cq, will be linear. These are called Stern-Volmer plots. It is important to point out that a linear Stern-Volmer plot can also be seen with Type I photochemistry. The only requirement for a linear Stern- Volmer plot is that there be an intermediate that can either be inactivated by the quencher or go on to produce the product being measured (6). The intermediate does not have to be singlet oxygen. Agreement between the slope of a Stern-Volmer plot of experimental data and the slope calculated from known singlet- oxygen quenching constants is strong evidence favoring a singlet-oxygen mediated mechanism.
Singlet-oxygen lifetimes in many solvents are known (24). Thus, Equation 19 can often be evaluated from data in the literature to calculate a theoretical value for the slope of a Stern-Volmer plot. Obtaining Stern-Volmer plots with more than one quencher will further improve the specificity of the method.
Deuterium-isotope solvent effect. The lifetime of singlet oxygen is considerably longer in solvents where deuterium has been substituted for hydrogen than the lifetime in unadulterated solvents (25). For example, the lifetime of singlet oxygen is 68 µs in deuterium oxide, but only 3.1 µs in water (26-27). Thus, under continuous irradiation, the steady-state concentration of singlet-oxygen should be higher in deuterated solvents and consequently, the rate of production of oxidation products should be higher. Thus, in very dilute solutions, the deuterium-isotope solvent effect is substantial. In dilute aqueous solutions, the deuterium-isotope solvent effect can be as large as 22.
However, caution is needed in interpreting the effect of deuterium substitution in the solvent, deuterium substitution can perturb photochemical systems in other ways. In some systems, the deuterium-substituted solvent will exchange deuterium atoms with hydrogen atoms in target molecules. The kinetics of a photooxidation being studied may then be changed due to primary or secondary kinetic isotope effects that can occur at any step in a complex photooxidation mechanism.
A primary kinetic isotope effect occurs when the deuterium-hydrogen exchange occurs at a bond that is either broken or formed during the oxidation. Primary kinetic isotope effects can be large. At 25° C, substitution of deuterium for hydrogen can reduce reaction rates by factors of up to 6.9, 9.2 and 11.5, for carbon-hydrogen bonds, nitrogen-hydrogen bonds and oxygen-hydrogen bonds, respectively (28). These ratios are comparable in magnitude to the ratios in singlet-oxygen lifetimes seen between deuterated solvents and non-deuterated solvents. Secondary kinetic isotope effects occur when the substitution of deuterium for hydrogen occurs at bonds that are not broken or made during the photooxidation. These effects are generally much smaller, but in water, the secondary solvent isotope effect can be large, due to changes in acidity (29). Substituting two deuterium atoms for two hydrogen atoms in the water molecule significantly changes its chemical properties (29).
When using the deuterium isotope solvent effect as a mechanistic test, it is best to select conditions where the reactions being studied go to completion. In some cases, this will reduce the perturbations caused by kinetic isotope effects. However, in systems, where two or more alternative reactions compete, kinetic isotope effects can still cause significant changes in the oxidation-product distribution even when the reactions are carried to completion.
The specificity of the deuterium-isotope solvent effect can be improved if one requires quantitative agreement between the amount of enhanced photooxidation and a theoretical value for the expected increase in singlet-oxygen lifetime. The expected increase in oxidation products caused by deuterium substitution in the solvent is given by Equation 20,
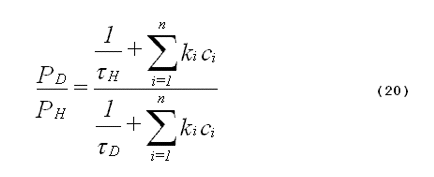
where PD is the amount of oxidation product formed in deuterated solvent, PH is the amount of oxidation product formed in non-deuterated solvent,
H is the
lifetime of singlet oxygen in non-deuterated solvent, D is the lifetime of singlet
oxygen in deuterated solvent, and n is the number of components in the system
other than the solvent. The definitions of the other symbols are the same as
those given in Equation 19. In Equation 20, we assume that no exchange of
deuterium for hydrogen occurs between the solvent and the substrates. The
numerator of Equation 20 represents the total singlet-oxygen quenching rate of
the system being studied using a non-deuterated solvent. The denominator
represents the total singlet-oxygen quenching rate for the system using the
analogous deuterated solvent.
The derivation of Equation 20 is analogous to the derivation of Equation 19. The student may wish to derive Equation 20 as an exercise.
From Equation 20, it follows that in some photochemical systems, the deuterium isotope solvent effect will be very small. This phenomenon is shown in Figure 7. When the rate of quenching of singlet oxygen by the various substrates present in the system is much greater than the rate of quenching by solvent, deuterium substitution in the solvent will not significantly affect the total quenching rate, and consequently there will be only a small effect on the lifetime of singlet oxygen.
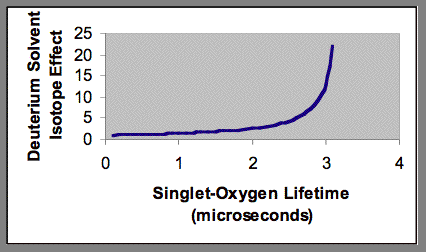
Figure 7. Blunting of the deuterium solvent effect by added quenching agents to an aqueous photochemical system. The singlet-oxygen lifetime shown on the x-axis is the result of quenching by both water and added quenching agents.
Substitution of deuterium oxide for water in cell cultures is particularly problematic. The effect of this solvent change on the complex biochemistry of cells is incompletely understood. Data from experiments of this type are particularly difficult to interpret when one uses a biological assay of cell damage, such as apoptosis, as opposed to a simple chemical assay, such as hydroperoxide formation. Further, there is uncertainty about the lifetime of singlet oxygen within the cell. Some studies suggest that the lifetime of singlet oxygen is less than 1 µs and that most of the singlet oxygen is quenched by the high concentration of biomolecules present within the cell rather than by water (30-33). If these studies are correct, one would expect a very small deuterium solvent effect. However, other studies suggest that the lifetime of singlet oxygen within the cell is close to that of water and that there is little quenching of singlet oxygen by biomolecules present within the cell (34). If these latter studies are correct, one would expect a very large deuterium solvent effect in cells.
Oxygen dependence. Photooxidations based on the generation of singlet oxygen are clearly oxygen dependent. In contrast, radical oxidations may or may not have an oxygen dependence. Very often free-radical chemistry is mediated by chain reactions, such as those shown by Equations 21 and 22.
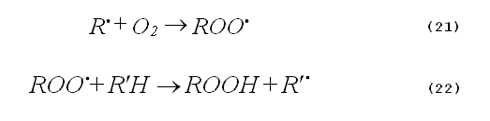
Assays for Free Radicals Including Superoxide Anion
Now we will consider the assays for the detection of radical-mediated photochemistry. Electron paramagnetic spin resonance (ESR) detection of spin-trap adducts is one of the most commonly used assays and is useful for most free radicals. Photochemistry that does not have a dependence on oxygen also provides strong evidence favoring free-radical mechanism. In some cases, time-resolved transient-absorption spectroscopy can be used to detect cationic or anionic photosensitizer radicals. Finally, cytochrome-c reduction that is inhibited by superoxide dismutase is a widely used assay for superoxide.
ESR spin trapping. Some free radicals have distinctive, narrow-lined ESR spectra and are relatively stable. Free radicals with these properties can often be directly detected by ESR. In photochemical oxidations, most free radicals of interest, such as hydroxyl radical or superoxide cannot be directly detected with ESR. Detection of these radicals depends on the use of spin traps (35-36). Spin traps are compounds that react with unstable radical species to form relatively stable radical adducts that have distinctive ESR spectra.
Most of the commonly used spin traps are nitrone or nitroso derivatives. One of the most popular spin traps for aqueous solutions is 5,5-dimethyl-1-pyrroline N- oxide (DMPO). This trap is useful for the identification of hydroxyl radicals, of various peroxyl radicals, and of superoxide.
Superoxide reacts with DMPO relatively slowly (35-36). As a result, very high concentrations of DMPO, on the order of 100 mM, are needed to efficiently trap superoxide in biological studies. These very high concentrations of DMPO can potentially be toxic to cells. The DMPO-OOH adduct can occasionally be confused with adducts formed from the reaction of peroxyl radicals with DMPO. As a consequence, definitive identification of superoxide requires that the DMPO-OOH adduct decrease with the addition of enzymatically-active superoxide dismutase, but not superoxide dismutase that has been denatured. An additional problem with the DMPO-OOH adduct is that it may decompose to produce the DMPO-OH adduct. Formation of this later adduct is usually taken as evidence for hydroxyl radicals.
Hydroxyl radicals react with DMPO to give the DMPO-OH adduct with a rate constant on the order of 109 M-1s-1. However, generation of the DMPO-OH adduct lacks specificity. The DMPO-OH adduct may result from the decomposition of the DMPO-OOH adduct or from the interaction of singlet oxygen with DMPO (37-38). Thus, additional studies are required to confirm the presence of free hydroxyl radicals. Generally, hydroxyl radical traps are used to carry out this confirmation. Ethanol, formate and dimethyl sulfoxide are common hydroxyl radical traps. These compounds react rapidly with hydroxyl radicals, successfully competing with DMPO. The products of the reactions of hydroxyl radicals with all of these three traps are carbon-centered radicals that subsequently may be trapped by DMPO. Thus, the addition of a hydroxyl-radical trap to the system should decrease the amount of DMPO-OH formed and replace it with the DMPO adduct of a carbon-centered radical. The specificity of the method is further improved if more than one hydroxyl-radical trap is used.
Oxygen dependence of products. As was discussed previously, the absence of oxygen dependence is a strong argument favoring radical-mediated photochemistry. In contrast, the presence of a dependence on the oxygen concentration has an ambiguous interpretation that can occur with both Type I and Type II photochemistry.
Time-resolved transient absorption spectroscopy. When charge is transferred from the photosensitizer to an acceptor molecule, either a cationic or anionic photosensitizer radical is produced. In some cases, these radical species have absorption spectra significantly different from the ground-state photosensitizer and from the excited triplet photosensitizer. In such cases, these radical species may be detected using time-resolved transient absorption spectroscopy (39). Generally the photosensitizer is excited with a pulsed laser. Time-dependent changes in the absorption spectra are then measured at right angles to the laser beam.
Superoxide-dismutase inhibitable reduction of cytochrome c.
This simple assay requires only a spectrometer (40-41). Cytochrome c is readily reduced by superoxide, but cytochrome c is also readily reduced by many other agents. Superoxide dismutase is an enzyme that specifically catalyzes the destruction of superoxide. The difference between the rate of reduction of cytochrome c in the presence of active superoxide dismutase and in the presence of denatured superoxide dismutase gives the rate of production of superoxide. Measurements of both DMPO spin trapping and cytochrome c reduction may be used together to provide additional confirmation of the production of superoxide.
Conclusions
Determining the mechanisms of photochemical oxidations has proven to be a challenging problem. A wide variety of assays are available for both singlet oxygen and for free radicals including superoxide. In photochemical oxidations, singlet-oxygen generation often competes with free-radical generation. The fraction of the phooxidation that proceeds by each route will depend upon the conditions. High oxygen concentrations favor the formation of singlet oxygen or of superoxide. In contrast, high target-molecule concentrations and low oxygen concentrations favor the formation of other free radicals.
References
1. Foote, C. S. (1991) Definition of type I and type II photosensitized oxidation. Photochem. Photobiol. 54, 659.
2. Vidòczy, T. (1992) Type I and Type II photosensitized reactions: reasons for dispute. J. Photochem. Photobiol. B: Biol. 14, 139-142.
3. Weldon, D., T. D. Poulsen, K. V. Mikkelsen, and P. R. Ogilby (1999) Singlet sigma: The "other" singlet oxygen in solution. Photochem. Photobiol. 70, 369- 379.
4. Foote, C. S. (1968) Mechanisms of photosensitized oxidation. Science 162, 963-970.
5. Foote, C. S. (1976) Photosensitized oxidation and Singlet-Oxygen: Consequences in Biological Systems. In: Free Radicals in Biology, Vol. II, Pryor, W. A., Ed., Academic Press, New York, pp. 85-133.
6. Foote, C. S., F. C. Shook and R. B. Abakerli (1984) Characterization of Singlet Oxygen. In: Oxygen Radicals in Biological Systems (Methods in Enzymology, Vol. 105), Packer, L., Ed., Academic Press, New York, pp. 36-47.
7. Laustriat, G. (1986) Molecular mechanisms of photosensitization. Biochim. 68, 771-778.
8. Foote, C. S. (1987) Type I and type II mechanisms of photodynamic action. In: Light-Activated Pesticides (ACS Symposium Series 339), J. R. Heitz and K. R. Downum, Eds., Am. Chem. Soc., Washington, pp. 22-38.
9. Moore, D. E. (1998) Mechanisms of photosensitization by phototoxic drugs. Mutation Res. 422, 165-173.
10. Tanielian, C., R. Mechin, R. Seghrouchni and C. Schweitzer (2000) Mechanistic and kinetic aspects of photosensitization in the presence of oxygen. Photochem. Photobiol. 71, 12-19.
11. Parker, J. G. and W. D. Stanbro (1984) Dependence of photosensitized singlet oxygen production on porphyrin structure and solvent. In Porphyrin Localization and Treatment of Tumors. Progress in Chemical & Biological Research Ser., Vol 170 (Edited by D. R. Doiron and C. J. Gomer), pp. 259-284, Alan R. Liss, New York.
12. Gorman, A. A., A. A. Krasnovsky and M. A. J. Rodgers (1991) Singlet oxygen infrared luminescence: unambiguous confirmation of a solvent-dependent radiative rate constant. J. Phys. Chem. 95, 598-601.
13. Wessels, J. M., and M. A. J. Rodgers (1995) Detection of
O2(1
g)-O2(3g) transition in aqueous environments: a Fourier-transform near-infrared
luminescence study. J. Phys. Chem. 99, 15725-15727.14. Kulig, M. J., and L. L. Smith (1973) Sterol metabolism. XXV. Cholesterol oxidation by singlet molecular oxygen. J. Org. Chem. 38, 3639-3642.
15. Korytowski, W., G. J. Bachowski and A. W. Girotti (1993) Analysis of
cholesterol and phospholipid hydroperoxides by high-performance liquid
chromatography with mercury drop electrochemical detection. Anal. Biochem.
213, 111-119.
16. Korytowski, W., G. J. Bachowski and A. W. Girotti (1991) Chromatographic
separation and electrochemical determination of cholesterol hydroperoxides
generated by photodynamic action. Anal. Biochem. 197, 149-156.
17. Smith, L. L., J. I. Teng, M. J. Kulig and F. I. Hill (1973) Sterol Metabolism.
XXIII. cholesterol oxidation by radiation-induced processes. J. Org. Chem.38,
1763-1765.
18. Yamamoto, Y., M. H. Brodsky, J. C. Baker and B. N. Ames (1987) Detection
and characterization of lipid hydroperoxides at picomole levels by high-
performance liquid chromatography. Anal. Biochem. 160, 7-13.
19. Bachowski, G. J., E. Ben-Hur and A. W. Girotti (1991) Phthalocyanine-
sensitized lipid peroxidation in cell membranes: use of cholesterol and azide as
probes of primary photochemistry. J. Photochem. Photobiol. B: Biol. 9, 307-321.
20. Vever-Bizet, C., M. Dellinger, D. Brault, M. Rougee, R. V. Bensasson (1989)
Singlet molecular oxygen quenching by saturated and unsaturated fatty-acids
and by cholesterol. Photochem. Photobiol. 50, 321-325.
21. Foote, C. S., R. B. Abakerli, R. L. Clough and R. I. Lehrer (1981) On the
question of singlet oxygen production in polymorphonuclear leucocytes. In:
Bioluminescence and Chemiluminescence, Basic Chemistry and Analytical
Applications, M. A. DeLuca and W. D. McElroy, Eds., Academic Press, New
York, pp. 81-88.
22. Steinbeck, M. J., A. U. Khan and M. J. Karnovsky (1992) Intracellular singlet
oxygen generation by phagocytosing neutrophils in response to particles coated
with a chemical trap. J. Biol. Chem. 267, 13425-13433.
23. Steinbeck, M. J., A. U. Khan and M. J. Karnovsky (1993) Extracellular
production of singlet oxygen by stimulated macrophages quantified using 9,10-diphenylanthracene and perylene in a polystyrene film. J. Biol. Chem. 268,
15649-15654.
24. Wilkinson, F., W. P. Helman, and A. B. Ross (1995) Rate constants for the decay and reactions of the lowest electronically excited singlet state of molecular oxygen in solution. an expanded
and revised compilation. J. Phys. Chem. Ref. Data 24, 663-1021.
25. Merkel, P. B., and D. R. Kearns (1972) Deuterium effects on singlet oxygen
lifetimes in solutions. a new test of singlet oxygen reactions. J. Am. Chem. Soc.
94, 1030-1031.
26. Egorov, S. Y., V. F. Kamalov, N. I. Koroteev, A. A. Krasnovskii, Jr., B. N.
Toleutaeu, and S. U. Zinukou (1989) Rise and decay kinetics of photosensitized
singlet oxygen luminescence in water. measurements with nanosecond time-
correlated single photon counting technique. Chem. Phys. Lett. 163, 421-424.
27. Ogilby, P. R., and Foote, C. S. (1982) Chemistry of singlet oxygen. 36. singlet
molecular oxygen (1g) luminescence in solution following pulsed laser
excitation. solvent deuterium isotope effects on the lifetime of singlet oxygen. J.
Am. Chem. Soc. 104, 2069-2070.
28. Hine, J. (1962) Physical Organic Chemistry, McGraw-Hill Book Company,
Inc., New York, pp. 71-73.
29. Hine, J. (1962) Physical Organic Chemistry, McGraw-Hill Book Company,
Inc., New York, pp. 120-121.
30. Matheson, I. B. C., R. D. Etheridge, N. R. Kratowich, and J. Lee (1975) The
quenching of amino acids and proteins. Photochem. Photobiol. 21, 165-171.
31. Moan, J., E. O. Pettersen and T. Christensen (1979) The mechanism of
photodynamic inactivation of human cells in vitro in the presence of
haematoporphyrin. Br. J. Cancer 39, 398-407.
32. Baker, A., and J. R. Kanofsky (1992) Quenching of singlet oxygen by
biomolecules from L1210 leukemia cells. Photochem. Photobiol. 55, 523-528.
33. Moan, J., and K. Berg (1991) The photodegradation of porphyrins in cells can
be used to estimate the lifetime of singlet oxygen. Photochem. Photobiol. 53,
549-553.
34. Skovsen, E., Snyder, J. W., Lambert, J. D., and Ogilby, P. R. (2005) Lifetime
and diffusion of singlet oxygen in a cell. J. Phys. Chem. 109, 8570-8573.
35. Buettner, G. R., and R. P. Mason (1990) Spin-trapping methods for detecting
superoxide and hydroxyl free radicals in vitro and in vivo. In: Oxygen Radicals in
Biological Systems, Part B, Oxygen Radicals and Antioxidants (Methods in
Enzymology, Vol. 186), Packer, L. and Glazer, A. N., Eds., Academic Press, Inc.,
San Diego, pp. 127-133.
36. Pou, S., D. J. Hassett, B. E. Britigan, M. S. Cohen and G. M. Rosen (1989)
Problems associated with spin trapping oxygen-centered free radicals in
biological systems. Anal. Biochem. 177, 1-6.
37. Feix, J. B., and B. Kalyanaraman (1991) Production of singlet oxygen-derived
hydroxyl radical adducts during merocyanine-540-mediated photosensitization:
analysis by ESR-spin trapping and HPLC with electrochemical detection. Arch.
Biochem. Biophys. 15, 43-51.
38. Bilsky, P., K. Reszka, M. Bilska and C. Chignell (1996) Oxidation of the spin
trap 5,5-dimethyl-1-pyrroline N-oxide by singlet oxygen in aqueous solution. J.
Am. Chem. Soc. 118, 1330-1338.
39. Lambert, C. R., and I. E. Kochevar (1997) Electron transfer quenching of the
rose bengal triplet state. Photochem. Photobiol. 66, 15-25.
40. Sanders, S. P., S. J. Harrison, P. Kuppusamy, J. T. Sylvester and J. L.
Zweier (1994) A comparative study of EPR spin trapping and cytochrome c
reduction techniques for the measurement of superoxide anions. Free Radic.
Biol. Med. 16, 753-761.
41. Viola, A., A. Jeunet, R. Decreau, M. Chanon and M. Julliard (1998) ESR
studies of a series of phthalocyanines. mechanism of phototoxicity. comparative
quantitation of O2-. using ESR spin-trapping and cytochrome c reduction
techniques. Free Radic. Res. 28, 517-532.
04/28/08
04/24/14