DINOFLAGELLATE BIOLUMINESCENCE
and its CIRCADIAN REGULATION
J. Woodland (Woody) Hastings
Deceased 08/06/14 [Obituary]
Dinoflagellates occur ubiquitously in the oceans as planktonic forms, and are largely responsible for the sparkling luminescence (often erroneously called phosphorescence) of the ocean, commonly seen at night (especially in summer) when the water is disturbed (Figure 1). They occur primarily in surface waters, and many species are photosynthetic. In the phosphorescent bays (e.g., in Puerto Rico), high densities of a single species (Pyrodinium bahamense) occur. Red tides are blooms, essentially monocultures of a given dinoflagellate species, which may or may not be luminous (Figure 2).

Figure 1. The artist Andrew Wyeth's rendition of dinoflagellate bioluminescence in the ocean as a poacher pulls a lobster trap at night.
When luminous species are abundant, one can see the undulating luminescent patterns of fish fleeing the approach of boats, and in their trailing wakes. World War II aviators based on aircraft carriers in the South Pacific tell of the ease with which they relocated their base ship after a mission, or equally well a target ship. A luminescent wake may extend for many kilometers behind a ship, as the persistent turbulence stimulates cells to emit light.

Figure 2. A red tide in the Yellow Sea off the coast of China.
About 6% of all dinoflagellate genera contain luminous species, but since there are no fresh water luminous species, the proportion of luminous forms in the ocean is even higher. As a group, dinoflagellates are important as symbionts, notably for contributing photosynthesis and carbon fixation in corals and mollusks. Unlike luminous bacteria, which serve as symbionts with many higher organisms, no luminous dinoflagellates are known to occur as symbionts.
Dinoflagellate luminescence may function as a protective mechanism by either or both of two mechanisms. Luminescent flashing is triggered by mechanical stimulation when predators such as crustacea are active. First, since the flash could startle or divert the predator, this may impede predation (Esaias and Curl, 1972). The response time to stimulation (msec) is certainly fast enough to function in this way. Alternatively, predators of the crustacea might be alerted by the flashing, and thereby reduce the number of crustacea. This is referred to as the burglar alarm hypothesis (Abrahams and Townsend, 1993).
Cell Biology
Luminescence in Lingulodinium polyedrum (formerly named Gonyaulax polyedra) is emitted from many (~400 cell-1) small (~0.5
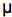 m) fluorescent organelles termed scintillons (Figure 3) (Nicolas et al., 1987). They occur as balloon-like out-pocketings of the cytoplasm into the cell vacuole with the neck remaining connected (Figure 4). They contain only dinoflagellate luciferase (LCF) and luciferin (dLH2), the latter associated with a separate protein, appropriately called luciferin binding protein (LBP). The number of scintillons changes with time of day, many being present during the night and few by day (Fritz et al., 1990). They can be identified by immunolabeling with antibodies raised against the luminescence proteins (Figures 4 and 5), and can be visualized by their bioluminescent flashing following stimulation, as well as by the fluorescence of luciferin (Figure 6) (Johnson et al., 1985).
m) fluorescent organelles termed scintillons (Figure 3) (Nicolas et al., 1987). They occur as balloon-like out-pocketings of the cytoplasm into the cell vacuole with the neck remaining connected (Figure 4). They contain only dinoflagellate luciferase (LCF) and luciferin (dLH2), the latter associated with a separate protein, appropriately called luciferin binding protein (LBP). The number of scintillons changes with time of day, many being present during the night and few by day (Fritz et al., 1990). They can be identified by immunolabeling with antibodies raised against the luminescence proteins (Figures 4 and 5), and can be visualized by their bioluminescent flashing following stimulation, as well as by the fluorescence of luciferin (Figure 6) (Johnson et al., 1985).
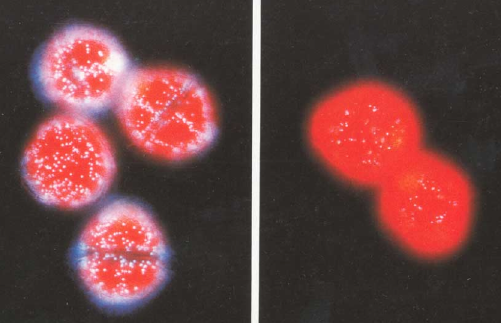
Figure 3. Fluorescence of luciferin in scintillons, seen as many small whitish spheres against the background red fluorescence of chlorophyll. There are many more in the cells during night (left) than day (right).
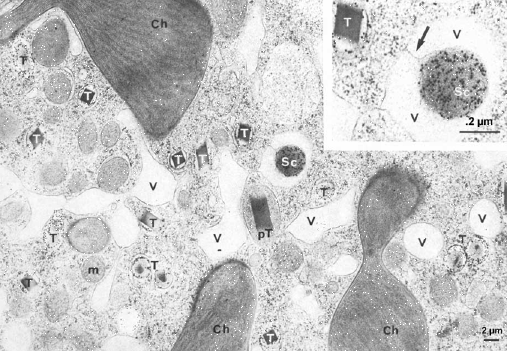
Figure 4. Gold labeled anti-luciferase marks a scintillon (Sc) hanging in a vacuole (V). Ch, chloroplast; M, mitochondrion; T, trichocyst; pT, pre-trichocyst.
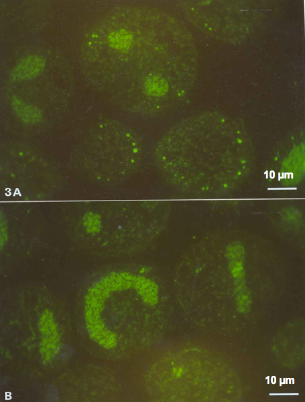
Figure 5. Lingulodinium scintillons labeled by anti-LBP at night (A) and day (B).
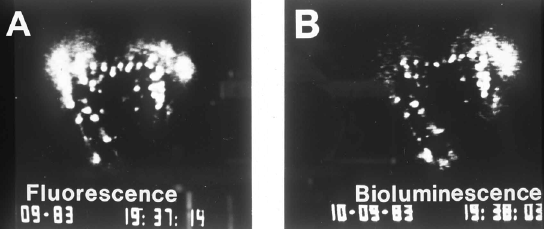
Figure 6. Co-localization of fluorescence (left panel) and bioluminescence (right) in a single Lingulodinium cell.
Light emission in dinoflagellates occurs in two different modes. Flashing, the display that is visible to the naked eye, consists of brief (0.1 sec) intense bursts of light (peak intensity, ~109 quanta sec-1 cell-1) (Figure 7). This strip chart recording of bioluminescence shows these flashes as unresolved vertical lines whose amplitudes are truncated by the slowness of the chart recorder pen. Cells also emit a glow, a low intensity emission that gradually rises to a peak (~104 quanta sec-1 cell-1), and then declines to zero over a period of several hours at the end of the night phase. The glow cannot be detected by eye, because it is masked by the brighter flashes, but is visible as a steady change in the baseline light emission on the chart recorder trace. It occurs at the end of night phase, and is believed to result from the daily breakdown of scintillons. It probably does not have any ecological significance. The total amount of light produced each daily cycle from the glow is about 107quanta cell-1.
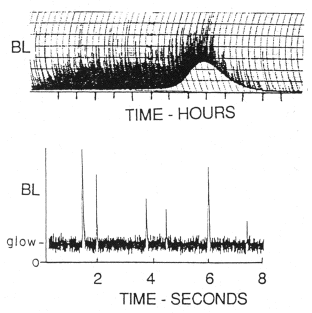
Figure 7. Glow and flashes on two time scales. Top panel: chart recording of bioluminescence (BL, with tics on the abscissa at hourly intervals) showing merged and truncated flashes superimposed on a glow that rises and falls at the end of the night phase. Bottom: Oscilloscope recording at a time when glow is being emitted, with flashes from single cells resolved.
Evolutionary Origins and Biochemistry
Luciferases are enzymes that catalyze reactions in light-emitting living organisms with substrates called luciferins (from the Latin, lucifer, light and fero, to bear, designated as LH2) There are many different luciferins and luciferases; for dinoflagellates, the luciferin is called dLH2 and the luciferase, LCF. The different systems have evidently originated and evolved independently, and the genes and proteins involved are unrelated to each other. It is not known how many times the property of light emission may have originated, since the loss of a luciferase system cannot be known from the geological record. There are, perhaps, 25 different extant luciferase systems (Hastings, 1983); major ones are listed in Table 1. For four of these systems, the structures of the luciferins and luciferases are known, along with the biochemistry of the reactions (Wilson and Hastings, 1998) (Table 2).

Table 2. Reaction schemes for four bioluminescent systems that use four different luciferases, showing the reactions of the different luciferins (with molecular oxygen in all cases) and the occurrence of peroxy intermediates.
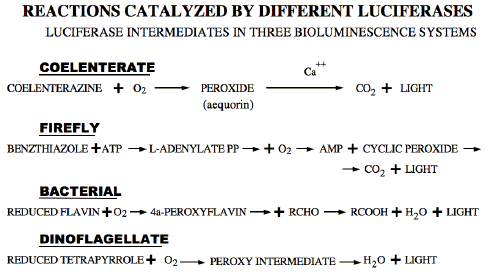
In spite of their differences, all such light emitting reactions have features in common, namely the reaction of the specific luciferin with oxygen to form a peroxide intermediate, whose breakdown populates an electronically excited state. In two known cases, cyclic peroxides are formed, which break down with the formation of carbon dioxide. In the other two, linear peroxide intermediates break down to give an excited product, designated as L=O, into which one atom of the oxygen molecule is incorporated.
Dinoflagellate luminescence appears to be of the latter type. Its luciferin is a novel reduced tetrapyrrole related to chlorophyll (Figure 8) (Nakamura et al., 1989). It has an absorption maximum at 390 nm and a fluorescence maximum at about 470 nm, matching that of the bioluminescence spectrum (Figure 9). The free product is non fluorescent; the identity of the emitter is non known. Luciferase is also unique, comprising in a single molecule three homologous domains, each with an active site, plus an N-terminal sequence of about 100 amino acids (Figure 10) (Li et al., 1997). The latter shares similarity to the N-terminal sequence of LBP. No genes with significant homology have been reported as of 2009, other than those in other luminous dinoflagellates.
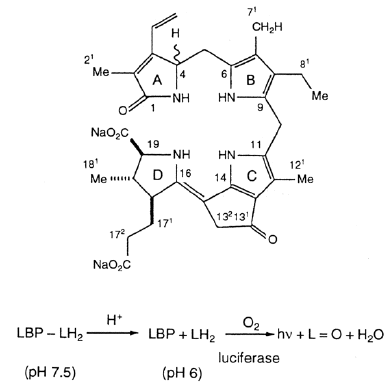
Figure 8. Dinoflagellate luciferin, dLH2, a reduced tetrapyrrole, isolated from Pyrocystis lunula.
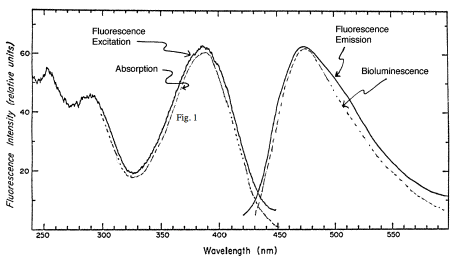
Figure 9. Absorption, fluorescence and bioluminescence spectra of dinoflagellate luciferin.
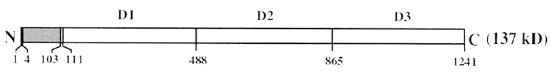
Figure 10. Structure of dinoflagellate luciferase with its three intramolecular homologous domains and N-terminal region.
Biochemistry and the Flashing Mechanism
Crude dinoflagellate extracts made at pH 8 do not emit light, but can be caused to do so by simply shifting the pH to about 6. Extracts are centrifuged to separate soluble from particulate components, including scintillons. Light emission occurs in both fractions, suggesting that during extraction some scintillons are lysed, releasing soluble luciferase and luciferin binding protein, while other scintillons seal off at the neck and form closed vesicles containing the luminescence proteins.
Gel filtration at pH 8 of the protein from the soluble extract results in a single peak of activity when fractions are assayed at pH 6.2 (Figure 11, top panel) (Fogel and Hastings, 1971). This is due to overlap in the elution of the two luminescence proteins, luciferase (~137 kDa) and LBP (~75 kDa), which can be purified separately. Active luciferin can be released from its binding protein by heating the pH 8 extract.
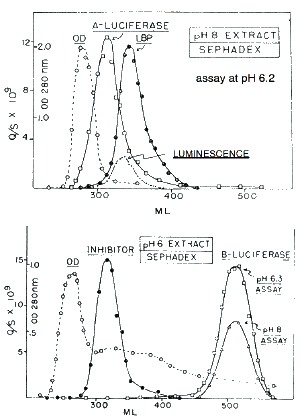
Figure 11. Sephadex filtration of extracts made at pH 8 (top) and pH 6 (bottom) See text for explanation.
A different picture is found after extraction at pH 6, where light emission occurs during extraction, so that all of the luciferin is used up. In the assays of fractions from gel filtration, free luciferin obtained separately must therefore be added to each to obtain activity. In this case, luciferase activity peaks in fractions corresponding to a much smaller size protein, about 35 kDa (Figure 11, bottom panel). This smaller size luciferase, referred to as B-luciferase (Krieger and Hastings, 1968), has activity at both pH 6 and pH 8, and the pH 8 activity (but not that at pH 6) is inhibited by a protein in the fractions with a molecular size corresponding to that of luciferin binding protein (Fogel and Hastings, 1971). As will be explained in more detail later, this B-luciferase is constituted of proteolytic fragments of luciferase, essentially single domains that lack an N-terminal regulatory domain, accounting for the activity at pH 8.
The reaction model thereby deduced for the full-length luciferase (and also complete single domains, but not B-luciferase), is that at pH 8 the luciferase is inactive and the luciferin is bound to luciferin binding protein (Figure 12, top), while at pH 6 luciferase becomes active and luciferin is released from the binding protein (Figure 12, bottom). The dependence of luciferase and LBP activities on pH, determined separately, is shown in Figure 13 (Morse et al., 1989a). The apparent inhibition by luciferin binding protein of the pH 8 activity of the 35 kDa B-luciferase occurs because luciferin binding protein binds luciferin, and makes it unavailable to the enzyme at that pH (Fogel and Hastings, 1971).
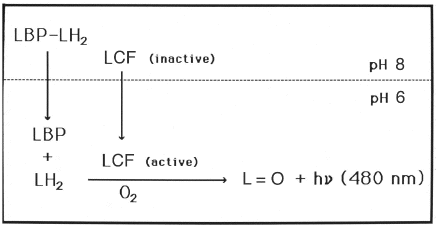
Figure 12. Activation of Lingulodinium bioluminescence upon a shift from pH 8 to 6 showing that luciferase changes from an inactive to active form while luciferin binding protein (LBP) releases bound luciferin.
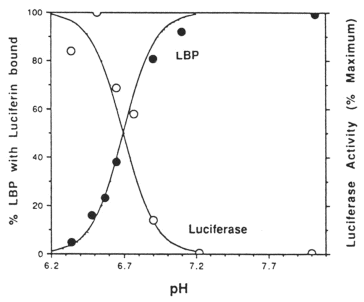
Figure 13. Effect of pH on the activities of Lingulodinium luciferase and LBP (luciferin binding protein).
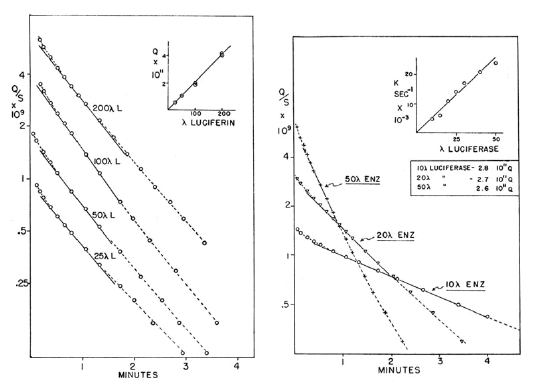
Figure 14. Kinetics of Lingulodinium luminescence reaction with varying concentrations of luciferin (left panel) and luciferase (right panel).
The kinetics of the reaction, determined at pH 6.4 and with B-luciferase (which has only a single catalytic site) and purified luciferin, show that the enzyme turns over with features characteristic of an enzymatic reaction. With a fixed concentration of luciferase and varying luciferin (Figure 14, left panel), the initial intensity and total light (inset of left panel) are proportional to luciferin concentration, and the rate constant for the reaction invariant. With fixed luciferin (right panel), the apparent rate constant is proportional to enzyme concentration (inset of right panel), and the total light invariant (Fogel and Hastings, 1971).
With isolated scintillons, the situation is completely different in that the kinetics are not dependent on concentration. This explains why flash kinetics in vivo are essentially the same under many different conditions (but not at different temperatures). The in vitro activity obtained by rapid (stopped-flow) mixing with acid to a final pH of 5.7 occurs as a flash with kinetics very close to those of the living cell (Figure 15).
Scintillon preparations can be purified by isopycnic density gradient centrifugation, either with sucrose (Figure 16) (Fogel and Hastings, 1972) or Percoll gradients (Desjardins and Morse, 1993). Following purification, scintillons can be visualized by their blue fluorescence (Figure 17), with some contaminating red-fluorescent chloroplast fragments evident. SDS-PAGE analysis showed only two major proteins with Mr values corresponding to luciferase and luciferin binding protein (Figure 18).
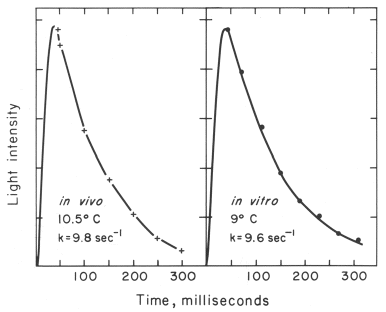
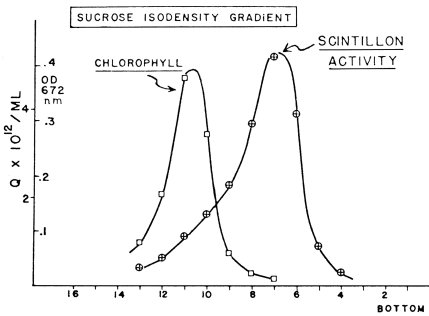
Figure 16. Scintillons band in a sucrose isodensity gradient and separate from chloroplasts. Abscissa: tube number.

Figure 17. Isolated Lingulodinium scintillons (blue fluorescence); Chloroplast fragments, red.
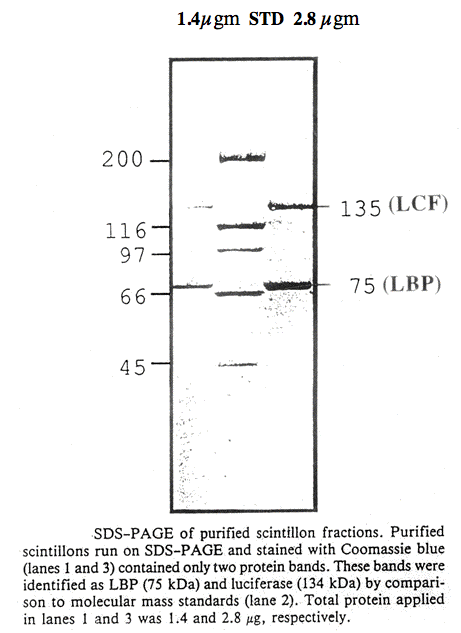
Figure 18. SDS-PAGE of purified scintillon fractions (De Jardins and Morse, 1993).
As mentioned above, the kinetics of the flash should be independent of the dilution of the suspension if the concentration of the enzyme does not change, which is so if it is contained in closed vesicles, with each vesicle acting as an independent unit. As shown in Figure 19, both initial intensity and total light are directly proportional to number of scintillons over five orders of magnitude. The integrity of the isolated scintillons remains after acidification and emission, such that additional flashes can be obtained by adjusting the pH back to 8 after a first flash, then incubating with purified free luciferin to produce recharged scintillons. Following another pH jump to pH 5.7, another flash, kinetically very similar to the first, occurs (Figure 20) (Fogel and Hastings, 1972). This recharging is not rapid and depends on both time and concentration of luciferin (Figure 21).
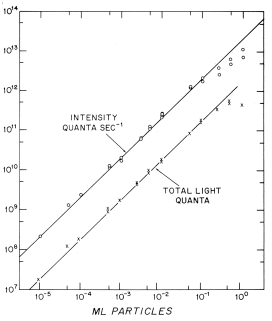
Figure 19. Relationship between concentration of scintillons and activity (both initial intensity and total light) over five orders of magnitude.
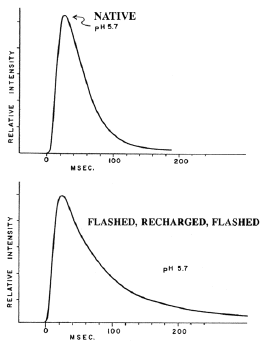
Figure 20. Kinetics of flashes of newly isolated scintillons (top panel), and recharged scintillons (bottom).
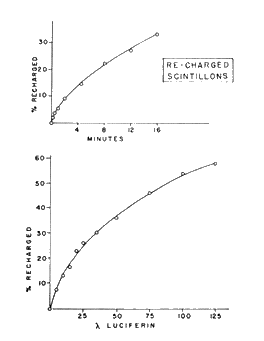
Figure 21. Time dependence (top panel) and effect of luciferin concentration (bottom) on recharging of scintillons.
Based on the results described above, the flashing of dinoflagellates in vivo was postulated to result from a transient pH change in the scintillons, triggered by an action potential in the vacuolar membrane initiated by membrane deformation. The action potential, while sweeping over the scintillons (Eckert, 1965), opens voltage gated ion channels, allowing protons from the acidic vacuole to enter, as modeled in Figure 22 (Fogel and Hastings, 1972; Nicolas et al., 1986).
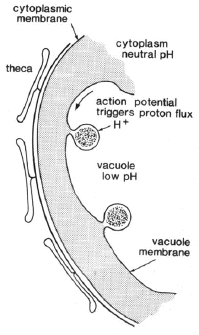
Figure 22. Schematic scintillons and the postulated mechanism for the pH-triggering of flashing.
Control of Dinoflagellate Luminescence: The Circadian Clock
The luminescence of G. polyedra and some other dinoflagellates is regulated by a cellular circadian biological clock mechanism, with the phase set by day-night, light-dark cycles (Morse et al., 1990; Hastings, 2001). The flashing response to mechanical stimulation is far greater during the night than during the day, and the low level spontaneous emission, a glow, rises and falls towards the end of the night phase (Figure 7). The circadian mechanism is endogenous. Cultures maintained under conditions of constant light and temperature can continue to exhibit rhythmically for weeks (Figure 23), but with a circadian period that is not exactly 24 hours. Like many other circadian processes, it is only approximately one day (from the Latin, circa, about and dies, day).
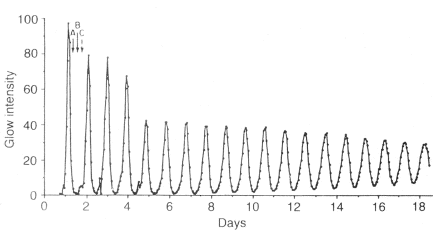
Figure 23. Persistent circadian rhythm of bioluminescence in a Lingulodinium culture maintained for 20 days in constant dim light. Other processes (A, B, C) also exhibit rhythms with maxima at different times of day, as indicated by arrows.
The biochemical and physiological nature of circadian clocks and genes involved are under intense investigation (Dunlap, 1999; Glossop and Hardin, 2002; Johnson, 2004). In humans and other higher animals, where it regulates the sleep-wake cycle and many other physiological processes, the mechanism involves the nervous system (Hastings et al., 1991; Zatz, 1992), but such rhythms also occur in plants and unicellular organisms, including Euglena, Chlamydomonas and Paramecium. In the case of Lingulodinium, daily changes occur in the cellular concentrations of luciferase, luciferin and its binding protein; remarkably, these and other proteins are synthesized and destroyed each day (Dunlap and Hastings, 1981; Johnson et al., 1984; Morse et al., 1989b; Marcovic et al., 1996). Indeed, and perhaps even more remarkably, the scintillons themselves are newly formed each night and disposed of in the early day phase (Figures 3 and 5) (Fritz et al., 1990). Chloroplasts are also remodeled on a daily basis (Rensing et al., 1980). Hence, the biological clock exerts control at a very basic level, by controlling gene expression protein synthesis and development.
The discovery that this circadian regulation occurs in Lingulodinium at the level of translation, as opposed to transcription, was not anticipated (Morse et al., 1989b; Hastings, 2001). The mRNAs for LBP and luciferase remain at the same levels over the circadian cycle, and many other proteins are circadian regulated (Milos et al., 1990: Marcovic et al., 1996). The mechanism has not been fully explained, but for LBP, which has been cloned and characterized (Lee et al., 1993), there is a novel 23 nucleotide UG repeat sequence in the 3'UTR of the mRNA where a potential regulatory protein might bind (Mittag et al., 1994). However, this sequence has not been found in any other Lingulodinium circadian-controlled mRNAs.
Evidence from several model circadian systems indicates that protein synthesis and protein phosphorylation are involved in the core biochemical oscillator. In Lingulodinium the strong phase-shifting by protein synthesis inhibitors and the effects of kinase and phosphatase inhibitors on period indicates that such steps are also of central importance in its core clock mechanism (Hastings, 2001).
The Three Luciferase Domains and Residues Involved in pH-activity Regulation
The full-length luciferase cDNA (4,037 base pairs) has an open reading frame of 3,723 base pairs, and encodes the 137 kDa luciferase protein (Li et al., 1997). It is comprised of three contiguous intramolecularly homologous domains, with no intervening nucleotides and no introns (Figure 24). When regions encompassing each of these were cloned and expressed separately, it was found that each is a catalytically active luciferase. At the amino acid level, the domains are ~ 75% identical overall, and ~95% identical in the more central regions (Figure 25), where the residues are presumed to be involved in catalysis.
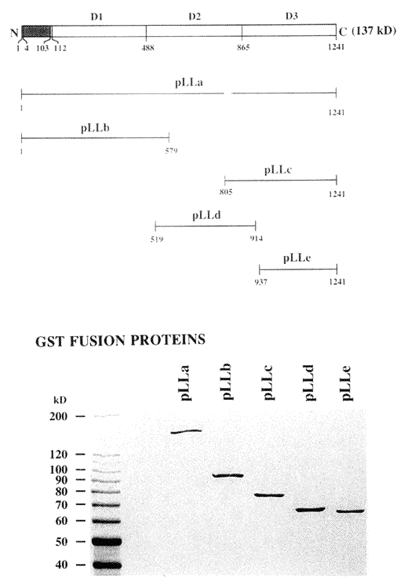
Figure 24. Luciferase structure and expression. Diagram showing the expression constructs containing full-length (pLLa) or partial sequences (pLLb, pLLc, pLLd and pLLe) of luciferase in relation to the full-length molecule, diagrammed at the very top, with the amino acid residue numbers of each peptide indicated. Below are shown Coomassie blue stained SDS-PAGE analyses of the expressed GST-luciferase fusion proteins, all of which are individually active.

Figure 25. Peptide sequence alignment of the three repeated domains, with the consensus residues shaded in black The first residue of each repeated domain is numbered 1.
In the more central regions, it is not only the amino acids that are conserved across the three domains; nucleotides are also conserved. A synonymous substitution is a nucleotide change in the DNA that does not change the amino acid in the protein for which it codes. Comparing the sequences of the three domains, there are very few synonymous substitutions in the central region, as compared to the two flanking parts of the domains (Li et al., 1997). Why would such nucleotide changes be less common in one region of the gene? A definitive explanation for this has not been obtained, but in another dinoflagellate, Pyrocystis lunula, the rate of synonymous substitution is the same in all regions of the domains (Figure 26) (Okamoto et al., 2001), so it must have some special function in Lingulodinium.
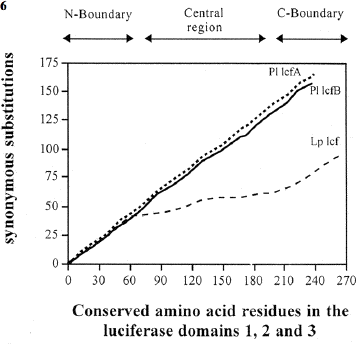
Figure 26. Frequency of synonymous substitutions along the domains of the P. lunula lcf (A, solid line, B, dashed line) and L. polyedrum lcf (bold-faced line) genes. The cumulative number of nucleotide substitutions was determined as related to the deduced amino acid sequences of the three repeat domains (D1, D2 and D3) of each luciferase gene.
The N-terminal region of luciferase (residues 4 to 103) shares a ~50% sequence similarity to the corresponding region of luciferin binding protein, suggesting that it serves a similar (but still unknown) function in the two proteins. It does not appear to be a signal or transit peptide, and luciferase and luciferin binding protein do not transit a membrane in the formation of the scintillons (Nicolas et al., 1987a; 1991). Also, the sequence is not required for the luminescence reaction, since the individual protein domains, which lack it, are active. It might be involved in the association between the two proteins. Scintillons are formed near the Golgi, where the two proteins associate before migrating together to the vacuolar membrane. But a similar sequence has been found to occur also in glucose S-transferase from P. lunula, a gene presumably not associated with bioluminescence, suggesting that it has a function in other systems.
The gene coding for the luciferase occurs in the genome as tandemly repeated copies and, like the lbp gene, does not contain introns (Li and Hastings, 1998). The spacer region between the lcf genes has no TATA box or other known promoter elements or consensus sequences. However, a conserved 13 nucleotide sequence, CGTGAACGCAGTG, which might be a dinoflagellate promoter, was identified by comparing the two intergene spacer regions of lcf, and the peridinin chlorophyll protein gene, pcp (Le et al., 1997).
We hypothesize that the three contiguous luciferase domains, each with its catalytic site and in association with luciferin binding protein, are structured within the scintillon as many supramolecular units. The presence of repeated conserved sequences in one enzyme molecule is not unprecedented, but to our knowledge, this is the only case in which each of the sequences has been shown to be separately active. A possible reason for the presence of three active sites on a single molecule is that it allows activity to be greater without an increase in the colloidal osmotic pressure of the scintillon.
At this point we can recall the very early observation, mentioned above, in which ~35 kDa fragments of the soluble luciferase were found to have activity, but that they lacked stringent pH control. They had activity at pH 8, whereas the full-length molecule did not. This activity can now be interpreted as coming from individual domains. Might the pH control be a property of the full-length molecule? The answer is no. Peptides embracing a single full domain exhibit pH activity curves that are the same as that of the full-length molecule (Figure 27). However, single domain peptides in which about 50 N-terminal amino acids are absent do exhibit higher activity at pH 8, like the proteolytic fragments. This is true for constructs of all three domains.
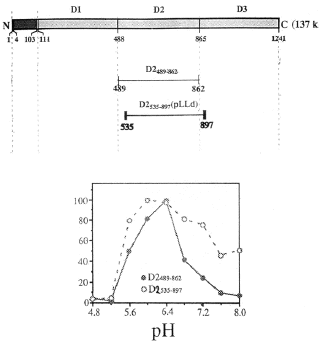
Figure 27. pH-activity curves of two peptides of domain 2, the full length D2 489-862 compared to the truncated D2 535-897, showing that the absence of amino acids from the N-terminal portion of the domain results in a greater luciferase activity in the alkaline region relative to that at the optimum. All curves are plotted normalized to the peak activities.
Inspection of the N-termini of the three domains reveals four conserved histidines (Figure 28; H1-4). One or more of these was postulated to be responsible for the low activity at pH 8. Indeed, replacement by site-directed mutagenesis of the first two by alanine results in luciferase activity at pH 8 nearly equal to that at pH 6.3, and replacement of three has an even greater effect (Figure 29) (Li et al., 2001). From this result and knowledge of the location of the histidines in the 3D structure of the domain, we have proposed that pH-initiated changes in charge states of these non-catalytic histidine residues regulate luciferase activity through conformational shifts in protein structure that allow luciferin to gain access to the active site
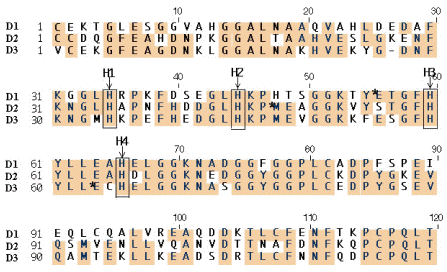
Figure 28. Amino acid alignment of N-terminal portion of the three luciferase domains, with boxes showing the four conserved histidines (H-1, H-2, H-3 and H-4) implicated in pH regulation of Lingulodinium luciferase.
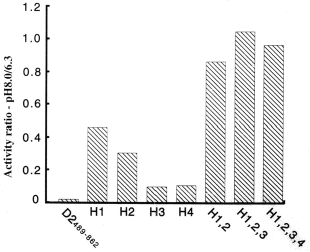
Figure 29. Replacement of conserved histidines by alanines, individually and together, increases activity at pH 8 relative to activity at pH 6.3 (ordinate).
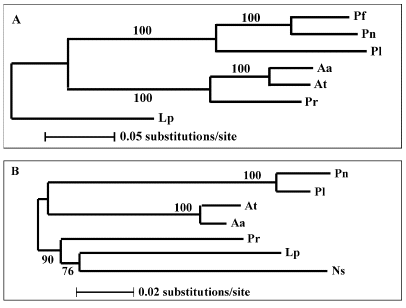
Figure 30. Trees depicting the molecular phylogeny of (A) dinoflagellate luciferases and (B) small subunit ribosomal RNAs based on nucleotide sequences. Numerals are the percentage of the bootstrap values.
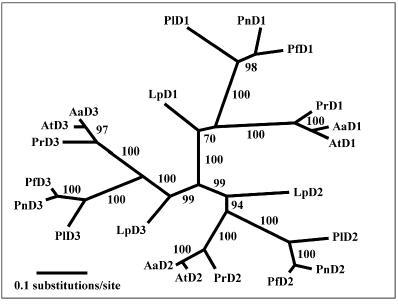
Figure 31. Tree of the three luciferase domains showing that the corresponding domains of the different individual species group together and that the domains of Lingulodinium separate early from those of the other species.
Luciferases From Other Dinoflagellate Species
There are numerous bioluminescent dinoflagellates, many of which have been brought into unialgal culture. The genes and proteins of seven such species have been examined, and the unusual three-domain structure of the gene occurs in all (Liu et al., 2004). Synonymous (but not non-synonymous) substitution rates in the central regions of the intramolecularly conserved domains vary considerably among the lineages, and are reduced severely in the Lingulodinium gene. The novel N-terminal sequence, which also occurs in the luciferin binding protein, also occurs in all, in spite of the fact that many of the species appear to lack luciferin binding protein or any other corresponding protein.
The luciferase tree based on the full-length coding sequences separates the seven genes into three clades (assemblages of species that all arose from a single common ancestor) (Figure 30A), as does the tree based on ribosomal RNA (Figure 30B). Both support the conclusion that the Gonyaulax (L. polyedrum) luciferase represents the most primitive form of the seven. In the ribosomal tree, a heterotrophic bioluminescent dinoflagellate, Noctiluca scintillans (Ns), represents the basal branch in dinoflagellates. Its luciferase can be expected to be even more primitive.
A tree constructed from each of the three individual domains (Figure 31), is congruent with the tree of the full-length coding sequence, suggesting co-evolution of three domains, which may be imposed by the requirement of the structural and functional coordination among the intramolecular domains during evolution. Indeed, individual domains are more similar to each other in the seven different species than they are to the other two domains within a species, indicating that the three domains evolved from an ancestral single luciferase domain structure and diverged for functional reasons.
As mentioned above, luciferase genes in both L. polyedrum and P. lunula were found to occur in tandem, and in 100 or more copies (Li and Hastings, 1998; Okamoto et al., 2001; Figure 32). It is possible, but not demonstrated that all copies may all be arrayed in tandem in the genome. The luciferase genes from five other species examined were found to be organized similarly, and the intergenic regions of all were determined (Figure 33) (Liu and Hastings, 2006).
While the sizes, coding sequences and repeat-domain structure of all are similar, the 5' UTR, 3' UTR and intergenic regions of the seven genes differ greatly in length and sequence, except for two stretches that are conserved in the intergenic regions of two pairs of phylogenetically-close species (Figure 33). Microsatellites and minisatellites were detected in the intergenic sequences of four species, Alexandrium affine, A. tamarense, Protoceratium reticulatum and Pyrocystis lunula, the first three of which have unusually high percentages of particular sets of dinucleotides. Most remarkably, the P. reticulatum intergenic region is almost exclusively made up of 19 nearly identical repeats of an 11-nucleotide sequence. Dinoflagellate luciferase intergenic regions have similarities to ribosomal genes and to some protein-encoding genes in trypanosomes, both of which are transcribed by RNA polymerase I. It is possible that the transcription of the dinoflagellate genes are catalyzed by an RNA polymerase with novel properties.
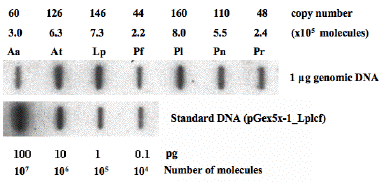
Figure 32. Determination of copy numbers of lcfs in seven species by slot-blot hybridization. After hybridization with radioactively labeled Lp lcf and analysis by densitometry (lower panel), various copy numbers (104 to 107) of Lp lcf cloned into pGEX 5X-1 (pGex5x-1_Lplcf) were loaded into the slots to generate a standard. The calculation of the copy numbers of lcfs of seven are based on the signal intensities that resulted from the hybridization of the Lp lcf with one microgram of genomic DNA of each species (upper panel). The genome sizes of all species are assumed the same and equal to 200 pg.
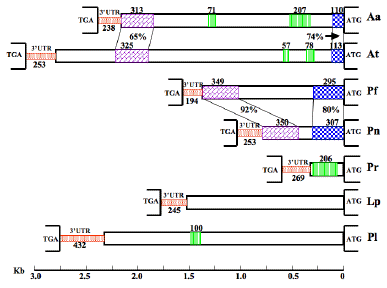
Figure 33. Intergenic regions of seven luciferases. TGA and ATG at left and right, respectively, define the beginning and end of the noncoding sequences (3'UTR, intergenic region and 5'UTR) between two tandem units, drawn in proportion to their lengths using the reference rule shown at the bottom of the diagram. 3'UTRs are represented by stippled narrower boxes, and the intergenic regions and 5'UTRs by wider ones. Blocks that show similarity between two species (Aa/At and Pf/Pn) are connected with pairs of lines, and have the identities shown. The regions with closely spaced vertical lines denote microsatellites or minisatellites, with the numbers indicating their lengths in nt.
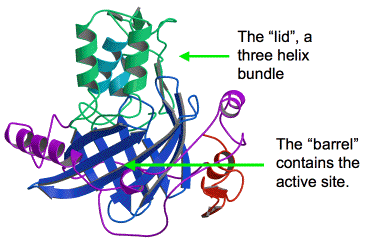
Figure 34. A schematic ribbon diagram of the crystal structure of luciferase domain 3 (D3). The barrel is composed of 10 anti-parallel beta strands (blue). The "lid" is composed of three regulatory alpha helices (green and cyan). The active site is located on the inside of the barrel, and is not accessible with the regulatory "lid" closed. Other features include a small C-terminal domain (red), and proline-rich sequences flanking the barrel (purple).
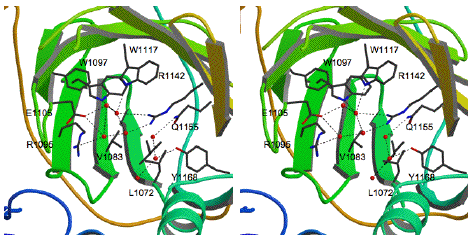
Figure 35. A stereo view of the interior of the luciferase barrel showing the arrangement of the amino acids that compose the active site. The amino acid side chains are shown as sticks (black) on a background of the beta strands (green). In this view, the regulatory helical bundle has been removed for clarity. Water molecules (red spheres) form hydrogen bonds (dotted lines) with active site residues. The water molecules occupy space in the active site that would be occupied by luciferin during the chemical reaction.
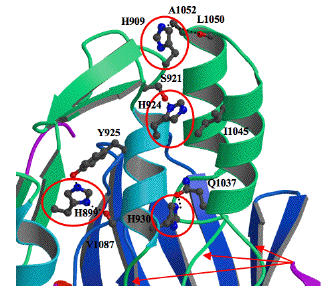
Figure 36. Enlarged view of the flexible regulatory three-helix bundle showing the location of the four histidines (red circles) and flexible gly-gly sequences (arrows at bottom). The histidine residues initiate a conformational change in the three-helix bundle that results in opening the active site. Three gly-gly sequences, G933-G934, G1035-1036 and G1068-1069, serve as hinges about which the chains move.
3-Dimensional Structure of Luciferase Domain 3
Enzymes have evolved to catalyze very specific chemical reactions. The three dimensional arrangement of amino acids within an enzyme determines what substrate is bound, what chemical reaction occurs, and what product is generated. The overall structure of an enzyme is created by folding the primary amino acid chain into a complicated three-dimensional conformation. During this process, the enzyme creates an active site, the specific arrangement of amino acids required to carry out the needed chemical reaction. The determination of an enzyme structure is therefore critical to provide a molecular picture of enzyme function.
The structure of the third domain (D3) of L. polyedrum luciferase was determined using X-ray crystallography, and refined to a resolution of 1.8 Å (Figure 34) (Schultz et al., 2005). The overall structure can be divided into two important parts: 1) the barrel-like structure that makes up the core of the enzyme and contains the active site where the oxidation of luciferin and light emission occur, and 2) the lid of the barrel, which is closed by a regulatory three-helix bundle at the top of the structure. The structure in Figure 34 was solved at pH 8, where the enzyme is inactive; the lid is closed and the luciferin substrate has no access to the active site on the inside of the barrel.
Although the structure at pH 6 is not known, molecular dynamics calculations indicate that the when the pH is drops to near pH 6, the regulatory N-terminal histidines initiate a conformation change in the three-helix bundle. The lid of the barrel opens and allows the luciferin substrate access to the active site, where it is oxidized. The interior of the barrel and arrangement of the active site residues are depicted in stereo in Figure 35. In this structure, water molecules are filling the active site that would be occupied by the luciferin substrate during the light producing chemical reaction.
As shown in Figure 36, the N-terminal histidine residues that regulate activity by pH are at the interface of the helices in the bundle. The pKa of histidine is usually in the range of 6 to 6.5, which indicates that the protonation state of these histidines is likely changing when the pH drops below 6.5, where the enzyme is active. Regulation of an enzyme reaction by pH is usually accomplished by altering the protonation state of an active site residue(s), making it favorable/unfavorable for a reaction to occur. For luciferase, the protonation state of histidine residues located outside the active site regulates the activity. This luciferase system is thus unique, using a pH-dependent structural change in the enzyme structure to regulate substrate access to the active site.
The 3-D structure of this luciferase is not only different from the other known luciferases (Figure 37; Table 2), its pH-dependent mechanism for activity regulation is different from that known for any enzyme, and nicely exemplifies convergent evolution where the same function can be achieved by different specific means.
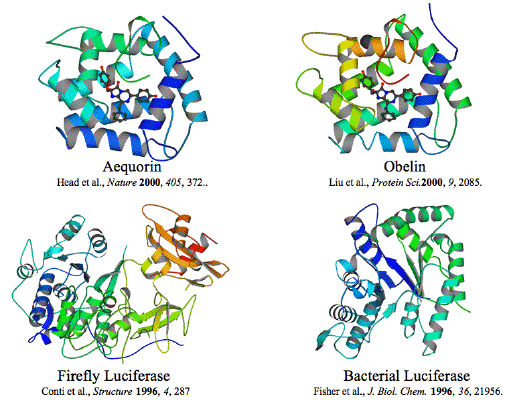
Figure 37. Structures of two coelenterate, a bacterial and a firefly luciferase. Note that the structures for the three phyla are very different, which reflects the different substrates and chemical reactions for each enzyme.
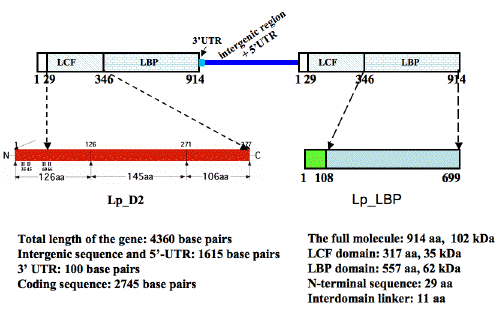
Figure 38. A schematic representation showing the genomic structure of Ns lcf genes and the domain organization of their predicted proteins in comparison with the second domain of Lp LCF and Lp LBP. At least two copies of the Ns lcf genes are tandemly arranged in a head-to-tail fashion, each consisting of 4360 bps. The precise boundary between the intergenic region and 5' UTR was not precisely mapped; together they account for 1518 bps. The 3' UTR of the Ns lcf is only 97 bps long, therefore much shorter than the 3' UTRs of lcf genes of seven photosynthetic dinoflagellates, which range from 194 to 432 bps. The largest open reading frame of Ns lcf predicts a protein of 914 amino acids with two major parts, an N-terminal luciferase-like (LCF-like) region and a C-terminal luciferin-binding protein-like (LBP-like) part. The LCF-like region shares a 56% sequence identity with, but is shorter at the N-terminus by 60 amino acid residues than, the individual domains of Lp LCF. The LBP-like part of 591 amino acid residues is about the same size as, and 41% identical with the comparable region of Lp LBP. No significant similarity was found between the N-terminal sequence of 29 amino acid residues of the Ns LCF, and any sequences in the database.
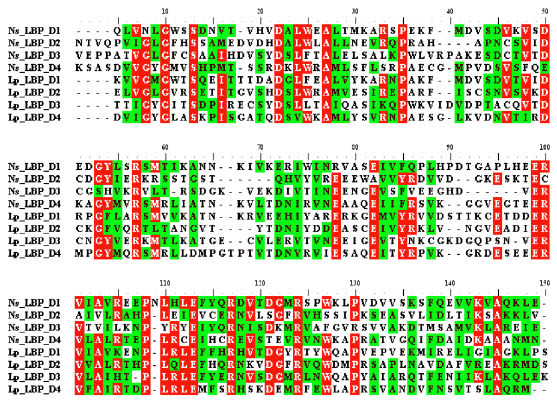
Figure 39. An alignment of the four internal repeats of Ns and Lp LBPs. The residues shaded or slightly shaded indicate that they are identical or conserved, respectively, in a majority of the sequences. Arrows mark the charged residues, which are present in all the sequences compared.
It was mentioned above that the luciferase of the more basal heterotrophic and unarmored bioluminescent dinoflagellate, Noctiluca scintillans, was expected to be more primitive, possibly having only a single luciferase domain. This has now been found to be so; the structural organization of its luciferase gene is strikingly different from that of the seven luminous species described above, all of which are photosynthetic (Liu and Hastings, 2007).
The Noctiluca gene codes for a polypeptide that consists of two distinct but contiguous regions (Figure 38). The N-terminal portion is shorter than, but similar in sequence to the individual domains of the three-domain luciferases found in all other luminous dinoflagellates studied. Thus, the catalytic component of its protein consists of only one luciferase domain.
The other part of the Noctiluca gene, situated at the C-terminal region, has size and sequence similarity to the luciferin-binding protein of Lingulodinium polyedrum (Gonyaulax polyedra), encoded there by a separate gene. In this study it was discovered that this gene and its protein are structured with domains, four in this case. Upon examination, the binding protein of L. polyedrum was discovered to also have a similar domain structure (Figure 39).
Western analysis shows that the native Noctiluca protein has the same size (~100 kDa) as the heterologously expressed polypeptide, indicating that it is not a polyprotein. Thus, sequences found in two proteins in the L. polyedrum bioluminescence system are present in a single polypeptide in Noctiluca.
Summary and Perspectives: Knowledge of One System Informs All Biology
Biological systems are marvelously diverse, and yet possess many similarities, sometimes obscured by additional features, and often attributable to evolutionary relatedness. In many cases, mechanisms that were deduced in one system are found to be important in others, where they might not have been readily appreciated. Thus, studies of the many diverse aspects of dinoflagellate bioluminescence can be viewed as leading to new discoveries, and an increased understanding of far more than photobiology, ranging across cell biology, cellular ultrastructure, molecular biology, gene regulation, biochemistry, enzyme structure, and molecular mechanisms.
Among the several novel findings in the dinoflagellate luminescence system is a new type of cell organelle, the scintillon, where the light emitting components are localized. With regard to luciferase, it is important to recall that domain 3 is only a part of the full-length protein, and that the luciferase itself is only one component of what must be a much larger multi-protein complex contained within the scintillons, including luciferin binding protein in at least some species. In producing the characteristic flash, the binding protein is presumably positioned favorably for the rapid transfer of luciferin to luciferase. Aside from membrane proteins, such as putative proton channels, scintillons contain only these two proteins. How luciferase and luciferin binding protein are arranged at the structural level in scintillons to accomplish the feat of flashing remains to be determined.
References
Abrahams M, Townsend L (1993) Bioluminescence in dinoflagellates: a test of the burglar alarm hypothesis. Ecology 74: 258-260.
Esaias W, Curl HJ (1972) Effect of dinoflagellate bioluminescence on copepod ingestion rates. Limnol Oceanogr 17: 901-906.
Desjardins, M and Morse, D (1993) The polypeptide components of scintillons, the bioluminescence organelles of the dinoflagellate Gonyaulax polyedra. Biochem Cell Biol 71, 176-82.
Dunlap, JC (1999) Molecular bases for circadian clocks. Cell 96: 271-290.
Dunlap, J and Hastings, JW (1981) The biological clock in Gonyaulax controls luciferase activity by regulating turnover. J Biol Chem 256: 10509-10518.
Eckert, Roger (1965) Biolelectric control of bioluminescence in the dinoflagellate Noctiluca. Science 147: 1140-1145.
Fogel, M and Hastings, JW (1971) A substrate binding protein in the Gonyaulax bioluminescence reaction. Arch Biochem Biophys 142: 310-321.
Fogel, M and Hastings, JW (1972) Bioluminescence: Mechanism and mode of control of scintillon activity. Proc Nat Acad Sci 69: 690-693.
Fritz, L, Morse, D and Hastings, JW (1990) The circadian bioluminescence rhythm of Gonyaulax is related to daily variations in the number of light emitting organelles. J Cell Science 95: 321-328.
Glossop, NR and Hardin, PE (2002) Central and peripheral circadian oscillator mechanisms in flies and mammals. J Cell Sci 115: 3369-3377.
Johnson, CH (2004) Precise circadian clocks in prokaryotic cyanobacteria. Curr Issues Mol Biol 6: 103-110.
Johnson, CH, Roeber, J, and Hastings, JW (1984) Circadian changes in enzyme concentration account for rhythm of enzyme activity in Gonyaulax. Science 223: 1428-1430.
Johnson, CH, Inoue, S, Flint, A and Hastings, JW (1985) Compartmentation of algal bioluminescence: autofluorescence of bioluminescent particles in the dinoflagellate Gonyaulax as studied with the image-intensified video microscopy and flow cytometry. J Cell Biol 100:1435-46.
Hastings, JW (1983) Biological diversity, chemical mechanisms and evolutionary origins of bioluminescent systems. J Molecular Evolution 19: 309-321.
Hastings, JW (2001) Cellular and molecular mechanisms of circadian regulation in the unicellular dinoflagellate Gonyaulax polyedra. In: Handbook of Behavioral Neurobiology,vol 12: Circadian Clocks (J Takahashi, F Turek & RY Moore, eds), pp 321-334, Plenum Press, NY.
Hastings, JW, Boulos, Z and Rusak, B (1991) Circadian Rhythms (CL Prosser, ed), In: Neural and Integrative Animal Physiology, pp 435-546 (Wiley Interscience, NY).
Krieger, N and Hastings, JW (1968) Bioluminescence: pH activity profiles of related luciferase fractions. Science 161: 586-589.
Le, QH, Markovic, P, Hastings, JW, Jovine, RVM and Morse, D (1997) Structure and organization of the peridinin chlorophyll protein gene in Gonyaulax polyedra. Mol Gen Genetics 255: 595-604.
Lee, D-H, Mittag, M, Sczekan, S, Morse, D and Hastings, JW (1993) Molecular cloning and genomic organization of a gene for luciferin-binding protein from the dinoflagellate Gonyaulax polyedra. J Biol Chem 268: 8842-8850.
Li, Liming, Hong, R and Hastings, J W (1997) Three functional luciferase domains in a single polypeptide chain. Proc Natl Acad Sci 94: 8954-8958.
Li, L and Hastings, JW (1998) The structure and organization of the Gonyaulax polyedra luciferase gene. Plant Molecular Biology 36: 275-284.
Li, Liming, Liu, Liyan, Hong, Robert, Robertson, Deborah and Hastings, J W (2001) N-terminal intramolecularly conserved histidines of three domains in Gonyaulax luciferase are responsible for loss of activity in the alkaline region. Biochemistry 40: 1844-1849.
Liu, L, Wilson, T and Hastings, J W (2004) Molecular evolution of dinoflagellate luciferases, enzymes with three catalytic domains in a single polypeptide. Proc Natl Acad Sci USA 101:16555-16560.
Liu, L and Hastings, J W (2006) Novel and rapidly diverging intergenic sequences between tandem repeats of the luciferase genes in seven dinoflagellate species. J Phycology 42:96-103.
Liu, L and Hastings, J W (2007) Two different domains of the luciferase gene in the heterotrophic dinoflagellate Noctiluca miliaris occur as two separate genes in photosynthetic species. Proc Natl Acad Sci USA 104:696-701.
Markovic P, Roenneberg T, Morse D (1996) Phased protein synthesis at several circadian times does not change protein levels in Gonyaulax. J Biol Rhythms 11:57-67.
Milos, P, Morse, D and Hastings, JW (1990) Circadian control over synthesis of many Gonyaulax proteins is at the translational level. Naturwissenschaften 77: 87-89.
Mittag, M, Lee, D-H, Hastings, JW (1994) Circadian expression of the luciferin-binding protein correlates with the binding of a protein to its 3' untranslated region. Proc Natl Acad Sci, 91: 5257-5261.
Morse, D, Pappenheimer, AM and Hastings, JW (1989a) Role of a luciferin binding protein in the circadian bioluminescent reaction of G. polyedra. J Biol Chem 264: 11822-11826.
Morse, D, Milos, PM, Roux, E, and Hastings, JW (1989b) Circadian regulation of the synthesis of substrate binding protein in the Gonyaulax bioluminescent system involves translational control. Proc Natl Acad Sci USA 86:172-176.
Morse, D, Fritz, L and Hastings, JW (1990) What is the clock? Translational regulation of circadian bioluminescence. Trends in Biochem 15: 262-265.
Nakamura, H, Kishi, Y, Shimomura, O, Morse, D, and Hastings, JW (1989) Structures of dinoflagellate luciferin and its enzymatic and non-enzymatic air-oxidation products. J Am Chem Soc 111: 7607-7611.
Nicolas, M-T, Nicolas, G, Johnson, CH, Bassot, J-M and Hastings, JW (1987) Characterization of the bioluminescent organelles in Gonyaulax polyedra (dinoflagellates) after fast-freeze fixation and antiluciferase immunogold staining. J Cell Biol 105: 723-735.
Nicolas, M-T, Morse, D Bassot, J-M, and Hastings, JW (1991) Colocalization of luciferin binding protein and luciferase to the scintillons of Gonyaulax polyedra revealed by immunolabeling after fast-freeze fixation. Protoplasma 160: 159-166.
Okamoto, O K, Liu, Liyun, Robertson, D L and Hastings, J Woodland (2001) Members of a dinoflagellate luciferase gene family differ in synonymous substitution rates. Biochemistry 40: 15862-15868.
Rensing, L, Taylor, W, Dunlap and Hastings, JW (1980) The effects of protein synthesis inhibitors on the Gonyaulax clock II: The effect of cyclohexamide on ultrastructural parameters. J Comp Physiol 138: 9-18.
Schultz, W, Liu, L, Cegielski, M and Hastings, JW (2005) Crystal structure of a pH-regulated luciferase catalyzing the bioluminescent oxidation of an open tetrapyrrole. Proc Natl Acad Sci USA 102: 1378-1373.
Wilson, T & Hastings, JW (1998) Bioluminescence. Annu Rev Cell Devel Biol 14: 197-230.
Zatz, M (Editor) (1992) Circadian Rhythms Discussions in Neuroscience VIII, Nos 2 + 3, pp 15-123.
02/05/09
A Pioneer in Bioluminescence Research
J. Woodland (Woody) Hastings, 87
Died 08/09/14
By Daniel E. Slotnik

His research led to the discovery of quorum sensing, the communicative power of bacteria. CreditDepartment of Molecular and Cellular Biology, Harvard University
J. Woodland Hastings, a Harvard biochemist whose improbable discovery of how bacteria communicate became the foundation for groundbreaking research in the development of more effective antibiotics, died on Wednesday at his home in Lexington, Mass. He was 87. The cause was pulmonary fibrosis, his family said.
Dr. Hastings devoted much of his career to studying bioluminescence, the light emitted by organisms like bacteria, fireflies and jellyfish. He was known for recognizing overarching biological processes in the humblest of organisms.
“One of Woody’s great fortes was coming up with concepts,” Ken Nealson, a microbiologist who worked with Dr. Hastings, said in an interview on Thursday. “He would see things other people wouldn’t see.”
In the late 1960s, Dr. Hastings and Dr. Nealson, then a postdoctoral fellow, noticed something curious about cultures of the luminescent marine bacterium Vibrio fischeri. (V. fischeri floats freely in the ocean and appears in greater concentrations in fish and squid.) The bacteria glowed intensely only after they reached a certain density — in a sense behaving like an army that waits until it has mustered enough troops to launch an attack. Dr. Hastings and Dr. Nealson surmised that the tiny organisms must be able to recognize the concentration of their fellows, probably through a signaling substance that they release and that then travels among them.
Researchers later theorized that the bacteria’s behavior was an evolutionary adaptation that would allow them to conserve energy until there were enough present to create a powerful glow. Once bacteria achieve a certain concentration, they can then take action en masse to fight an infection, say, or, conversely, cause one.
The idea that bacteria can communicate across cell barriers was initially met with skepticism. Dr. Nealson, now an environmental science professor at the University of Southern California, said editors of scientific journals would say: “ ‘We can’t find anything wrong with this paper, but it’s absurd. Bacteria don’t do this.’ ”
“I think I would have given in to the criticism,” Dr. Nealson added. “Woody had a lot more experience and was tougher than me.”
Their theory on the communicative power of bacteria, which came to be called quorum sensing, was not widely accepted until researchers, in the mid-1980s, identified the molecule that allows V. fischeri to communicate and the process by which it stimulates illumination. And it was not until the 1990s that scientists recognized how common quorum sensing is among various kinds of bacteria. Variations of the process appear in innumerable bacteria, from the benign V. fischeri to potentially deadly pathogens like E. coli.
Researchers are now investigating a variety of applications based on the idea, like antibiotics that could disrupt a specific infection without otherwise harming the host.
One area of interest is impeding the formation of biofilms. Biofilms, dense coagulations of bacteria like dental plaque, are responsible for an estimated 65 percent of bacterial infections and make bacteria more resistant to antibiotics. Drugs designed to silence communications among germs could theoretically keep them from massing.
John Woodland Hastings was born to Vaughan and Katherine Hastings in Salisbury, Md., on March 24, 1927. He left home at 10 to attend choir school at the Cathedral of St. John the Divine in Manhattan, then attended the Lenox School for Boys in Massachusetts on a scholarship. He received a bachelor’s degree in biology from Swarthmore College in 1947 and his master’s and doctorate from Princeton University. While at Princeton, he studied bioluminescence with E. Newton Harvey, a leader in the field, and became fascinated.
Over the next two years, on a postdoctorate fellowship at Johns Hopkins University, he studied the biomechanical process that makes fireflies glow. He paid children a penny per firefly to help gather enough material.
In 1953, he married Hanna Machlup and accepted his first faculty position at Northwestern University. While there, he began studying thedinoflagellate Gonyaulax polyedra, a plankton that flashes at night when stimulated. With Beatrice M. Sweeney, a researcher at the Scripps Institution of Oceanography in San Diego, he showed that the dinoflagellates operate on an internal biological rhythm, their flashes based on a circadian, or 24-hour, cycle.
It is now accepted that virtually every living creature operates on an internal circadian cycle. Dr. Hastings’s research also helped lay the foundation for treating some sleep disorders. Dr. Hastings accepted a position at the University of Illinois at Urbana-Champaign in 1957 and moved to Harvard in 1966. He also conducted research at the Marine Biological Laboratory at Woods Hole for more than five decades and served as a trustee there from 1967 until 1974. He retired about five years ago.
In 2003, he was inducted into the National Academy of Sciences. He and a colleague, Thèrése Wilson, wrote “Bioluminescence: Living Lights, Lights for Living” (2013). Dr. Hastings is survived by three daughters, Marissa Bingham, Jennifer Hastings and Laura Hastings; a son, David; his companion, Barbara Cheresh; a sister, Anne MacQueen; and five grandchildren. His wife died in 2009.
Dr. Hastings was more interested in advancing scientific understanding than practical applications of his work.
“Working on topics such as bioluminescence or circadian rhythms could only be motivated by a true interest in basic knowledge,” he said in “Fifty Years of Fun,” a lecture about his career. “It’s not leading directly to a solution for cancer.”
A version of this article appears in print on August 10, 2014, on page A20 of the New York edition of the New York Times with the headline: J. W. Hastings, 87, a Pioneer in Bioluminescence Research.